
記住我
Graphical Abstract.
IntroductionFrancisella is a genus of gram-negative bacteria that includes the highly virulent species F. tularensis, known for causing tularemia in humans and other mammals. Researches have revealed that F. tularensis can evade the host’s immune response by escaping the phagosome. Instead of being targeted for degradation, the bacterium can replicate within the host cell’s cytoplasm, where it is protected from immune responses. One intriguing aspect of the interaction between F. tularensis and its host cells is its ability to dampen the host’s cellular processes, including autophagy. The bacterium has developed mechanisms to manipulate the host cell’s autophagy machinery promoting its intracellular survival. Studies have shown that F. tularensis can inhibit the fusion of autophagosomes with lysosomes, where the contents are typically degraded. By preventing this fusion, the bacterium can avoid destruction and create a favorable intracellular niche for replication. Understanding the interplay between F. tularensis and autophagy is crucial for developing more effective treatments and vaccines against tularemia.
The basics of autophagyAutophagy is a fundamental cellular process responsible for the degradation and recycling of cellular components. This mechanism is crucial for maintaining cellular homeostasis, responding to stress conditions, and survival during nutrient deprivation. Autophagy involves the formation of autophagosomes, double-membraned vesicles that engulf damaged organelles, misfolded proteins, and pathogens for degradation in the lysosomes (Levine and Kroemer, 2019).
The process is highly regulated and can be triggered by various cellular signals, including nutrient levels, growth factors, and intracellular energy status. The regulation of autophagy is mediated by a complex network of signaling pathways, with the mammalian target of rapamycin (mTOR) pathway playing a central role in inhibiting autophagy under nutrient-rich conditions. Conversely, activation of AMP-activated protein kinase (AMPK) promotes autophagy by inhibiting mTOR under nutrient scarcity (Kim et al., 2011).
Autophagy not only helps in the removal of cellular debris but also plays a crucial role in defense mechanisms against infections by degrading intracellular pathogens through a process known as xenophagy (Deretic et al., 2013). Nevertheless, some pathogens together with F. tularensis can affect host cellular processes, including autophagy, to their advantage.
Mechanisms and pathwaysAutophagy is a cellular degradation process crucial for maintaining cellular homeostasis by removing damaged organelles and misfolded proteins. There are three primary types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), each with distinct mechanisms and functions.
Macroautophagy is the most extensively studied form of autophagy and involves the formation of double-membrane vesicles called autophagosomes. These vesicles engulf cytoplasmic material and then fuse with lysosomes, where the content is degraded and recycled. The process begins with the nucleation and expansion of the isolation membrane, which requires the expression of various autophagy-related proteins (ATGs). Key regulators of this pathway include the ULK1 complex, Beclin-1, and the ATG8/LC3 family proteins. This type of autophagy is highly regulated by nutrient availability and cellular stress, playing a critical role in cell survival during starvation and other stress conditions (Yamamoto and Matsui, 2024).
One of the primary roles of macroautophagy in host defense is the direct elimination of intracellular pathogens. This process, termed xenophagy, targets bacteria, viruses, and other pathogens for degradation. Xenophagy, also known as selective autophagy of pathogens, is a host defense mechanism where intracellular pathogens are recognized, sequestered within double-membraned autophagosomes, and subsequently degraded after fusion with lysosomes. This process is essential for maintaining cellular homeostasis and protecting the host from infections by targeting and eliminating invasive microbes such as bacteria, viruses, and parasites (Yuk et al., 2012). Xenophagy not only removes these pathogens but also facilitates antigen presentation, enhancing the adaptive immune response. The process of xenophagy begins with the recognition of intracellular pathogens through pattern recognition receptors (PRRs) that identify pathogen-associated molecular patterns (PAMPs). By delivering pathogen-derived antigens to major histocompatibility complex (MHC) molecules, autophagy enhances the presentation of these antigens to T cells. This process is critical for the immune system to recognize and respond to infections effectively (Van Kaer et al., 2019). Autophagy contributes to the presentation of viral antigens on MHC class II molecules, thus aiding in the activation of CD4+ T cells during viral infections (Schmid et al., 2007). Once recognized, adaptor proteins such as p62/SQSTM1 link the pathogen to autophagic machinery by binding to ubiquitinated microbial proteins and recruiting autophagy-related proteins like LC3 (Zheng et al., 2009). This recruitment ensures the encapsulation of the pathogen within autophagosomes. However, many pathogens have evolved sophisticated strategies to evade or manipulate xenophagy. Some escape recognition by autophagic receptors, while others inhibit autophagosome formation or prevent their fusion with lysosomes (Mao and Klionsky, 2016).
In addition to direct targeting pathogens, autophagy plays a significant role in modulating inflammatory responses. By degrading damaged organelles and excess proteins, autophagy prevents the accumulation of cellular debris that can trigger inflammation. Furthermore, autophagy regulates the production of pro-inflammatory cytokines, such as IL-1β, by controlling the activation of the inflammasome (Harris et al., 2011; Claude-Taupin et al., 2018; Iula et al., 2018).
Microautophagy involves the direct engulfment of cytoplasmic material by the lysosome itself through invagination, protrusion, or septation of the lysosomal membrane. Unlike macroautophagy, this process does not require the formation of autophagosomes. Instead, the lysosomal membrane engulfs small portions of the cytoplasm, including organelles and proteins, which are then degraded within the lysosome. Microautophagy is less well understood than macroautophagy but is believed to be crucial for maintaining organelle size and number, as well as for responding to nutrient depletion (Yamamoto and Matsui, 2024).
Chaperone-mediated autophagy (CMA) is distinct from both macroautophagy and microautophagy since it specifically targets soluble cytosolic proteins for degradation. This selectivity is mediated by chaperone proteins, particularly heat shock protein 70 (Hsp70), which recognize and bind to specific KFERQ-like motifs in the substrate proteins. These substrates are then translocated across the lysosomal membrane via the lysosomal-associated membrane protein 2A (LAMP-2A). CMA is involved in the degradation of long-lived proteins and plays a significant role in cellular quality control and stress response (Orenstein and Cuervo, 2010; Kaushik and Cuervo, 2018).
Pathogenic bacteria and their influence on autophagySome pathogenic bacteria inhibit autophagy to avoid degradation. They employ various strategies to overcome autophagy, such as inhibiting autophagosome formation, evading recognition, and preventing acidification. This section summarizes several bacterial pathogens and their effectors (see Table 1).
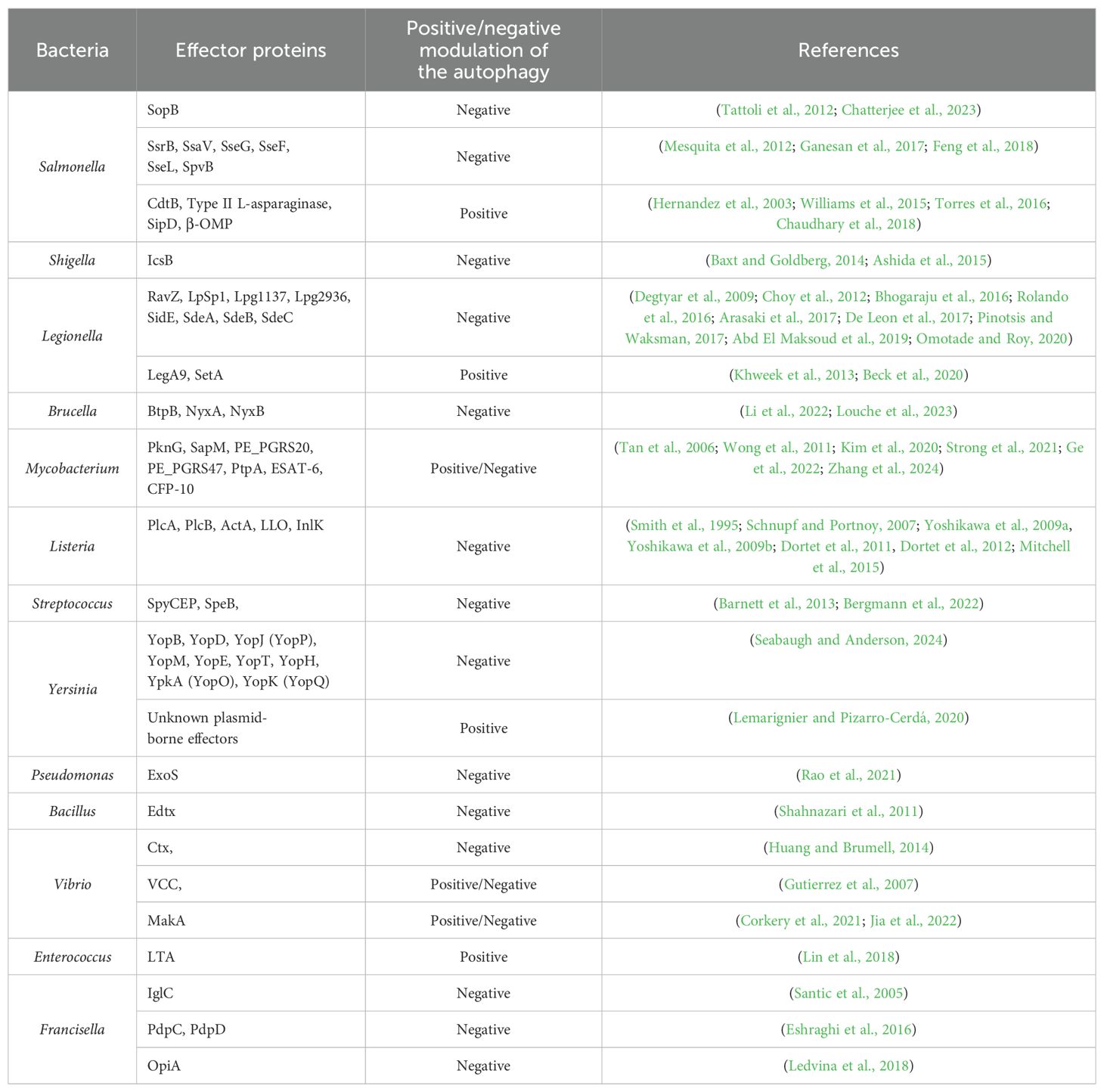
Table 1. Bacteria and their effector proteins affecting autophagy.
For instance, Salmonella enterica serovar Typhimurium, a common cause of foodborne illness, secretes effector proteins through its type III secretion system (T3SS) to inhibit autophagy. The bacterial effector protein SopB disrupts the formation of autophagosomes by interfering with the host cell’s signaling pathways (Tattoli et al., 2012; Chatterjee et al., 2023). This inhibition allows Salmonella to reside within a modified vacuole, where it can replicate and evade host immune responses. Other studies have also confirmed that effectors of SPI-2 (Salmonella pathogenicity island 2) T3SS as well as Salmonella virulence plasmids are important for bacterium to escape phagocytosis (Wu et al., 2020). For example, SsrB and SsaV effector proteins are responsible for activating mTOR through disruption of AMPK signaling (Ganesan et al., 2017). Other effector proteins, SseG and SseF, block Rab1 activity in host cell, which results in reduction of autophagosome formation and effective bacterial replication in cytoplasm (Feng et al., 2018). SseL acts as a deubiquitinase, which removes ubiquitin markers from Salmonella-infected cells, thus enabling bacterial replication instead of autophagic degradation (Mesquita et al., 2012). On the other hand, there exist Salmonella´s proteins that are involved in autophagy induction. L-asparaginase hydrolyzes the L-asparagine, thus T cells are not activated, which results in mTOR signaling inhibition and autophagy induction (Torres et al., 2016). Among other autophagy inducing proteins belong β-barrel outer membrane protein (β-OMP), Salmonella invasive protein D (SipD) and cytoskelethal distending toxin B (CdtB) (Hernandez et al., 2003; Williams et al., 2015; Chaudhary et al., 2018).
Shigella, a genus of bacteria responsible for causing dysentery, has developed sophisticated mechanisms to evade and manipulate the host’s immune responses, particularly autophagy (Phalipon and Sansonetti, 2007). Shigella can inhibit the formation of autophagosomes, the vesicles responsible for sequestering pathogens. This inhibition prevents the bacteria from being engulfed and degraded. Shigella achieves this through the secretion of effector proteins via its T3SS, which interfere with autophagy signaling pathways. Even if Shigella is initially captured by autophagosomes, it can escape before being degraded. The effector protein IcsB, secreted by Shigella, helps it evade autophagic recognition. IcsB interferes with the recruitment of autophagy-related proteins, such as ATG5, which are necessary for the formation of autophagosomes (Baxt and Goldberg, 2014; Ashida et al., 2015).
Legionella pneumophila, the causative agent of Legionnaires’ disease, hijacks the autophagic machinery through the irreversibly inactivating ATG8 proteins to create a replicative niche known as the Legionella-containing vacuole (LCV). Legionella secretes many effector proteins (Thomas et al., 2020), such as RavZ, that modulate autophagy to divert autophagic vesicles away from the lysosomal degradation pathway, thereby creating a favorable environment for bacterial replication (Choy et al., 2012).
LpSpl, a Legionella effector, mimics the enzymatic activity of sphingosine-1-phosphate lyase 1 (SGPL1), a key regulator of sphingolipid metabolism. LpSpl’s localization to mitochondria and the endoplasmic reticulum suggests a complex interplay with host metabolic pathways (Degtyar et al., 2009; Rolando et al., 2016). By reducing sphingosine levels, which are known to stimulate autophagy (Dall’Armi et al., 2013), LpSpl inhibits autophagosome biogenesis. This inhibition is dependent on its enzymatic activity, as mutants lacking functional active sites do not affect autophagy (Rolando et al., 2016). Despite its ability to modulate autophagy, LpSpl does not significantly impact bacterial replication in macrophages or amoebae but is required for optimal replication in mouse models (Rolando et al., 2016).
Lpg1137, a serine protease, disrupts autophagosome biogenesis by degrading Syntaxin 17 (Stx17), a key SNARE protein (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) involved in autophagosome-lysosome fusion and autophagy initiation (Hamasaki et al., 2013; Kumar et al., 2019). By cleaving Stx17, Lpg1137 prevents the formation of LC3- and ATG14-positive puncta, essential for autophagosome formation (Arasaki et al., 2017). Interestingly, bioinformatic analysis suggests Lpg1137 resembles mitochondrial SLC25 carrier proteins, though its exact mechanism requires further structural characterization (Gradowski and Pawłowski, 2017).
The effector Lpg2936 modulates host autophagy by inducing epigenetic changes in the promoter regions of key autophagy genes, such as ATG7 and LC3B. By methylating the GATC motif in these promoters, Lpg2936 suppresses autophagosome formation, indirectly promoting bacterial replication (Abd El Maksoud et al., 2019). While this highlights its role as a transcriptional regulator, its potential autophagy-independent roles remain under investigation (Pinotsis and Waksman, 2017).
In contrast to other effectors, LegA9 enhances the recruitment of SQSTM-1 to Legionella-containing vacuoles (LCVs), promoting their recognition for autophagic clearance (Khweek et al., 2013). However, this effector does not directly activate autophagy, and its role appears to be more relevant in alternative hosts, suggesting an evolutionary trade-off in mammalian infections (Price et al., 2020).
The SidE family of effectors (SidE, SdeA, SdeB, and SdeC) inhibits the recruitment of autophagy adapters such as SQSTM-1 to LCVs by generating unique ubiquitin linkages (Bhogaraju et al., 2016; Omotade and Roy, 2020). Furthermore, these effectors promote TFEB (transcription factor EB) nuclear translocation, which upregulates autophagy and lysosomal genes, suggesting a dual role in balancing nutrient acquisition and autophagy inhibition (De Leon et al., 2017). Temporal regulation appears essential, as SidE activity is blocked during co-expression with other Legionella effectors (De Leon et al., 2017).
SetA glucosylates TFEB, preventing its cytoplasmic retention and promoting its nuclear translocation, thereby inducing autophagic gene expression (Beck et al., 2020).
Brucella abortus, a pathogen responsible for brucellosis, resides in a membrane-bound compartment called the Brucella-containing vacuole (BCV) upon entry into host cells (Celli, 2019). Brucella inhibits the fusion of intermediate BCVs with late endosomes and lysosomes, which are critical steps in the autophagic process. Despite preventing full fusion with lysosomes, BCVs acquire several markers of late endosomes, including Rab7, a small GTPase, and its effector Rab-interacting lysosomal protein (RILP). This interaction allows Brucella to manipulate the endocytic pathway while avoiding degradation (Starr et al., 2008). Brucella effector like BtpB interferes with the host’s autophagic signaling pathways, allowing the bacteria to persist in a replication-permissive environment (Li et al., 2022). Interestingly, some bacteria can both inhibit and exploit autophagy at different stages of infection. The Brucella effector proteins NyxA and NyxB modulate host autophagy by targeting the SUMO-specific protease SENP3, which regulates autophagy-related protein de-SUMOylation. By disrupting SENP3 activity, NyxA and NyxB inhibit xenophagy, allowing Brucella to evade autophagic degradation and establish a replicative niche. This interference also dampens inflammatory responses associated with autophagy, promoting bacterial survival and persistence within host cells (Louche et al., 2023).
Mycobacterium tuberculosis, the bacterium responsible for tuberculosis, initially inhibits autophagy to prevent its destruction within macrophages. However, during later stages of infection, M. tuberculosis can exploit the autophagic process to access nutrients and enhance its survival. This dual manipulation underscores the complexity of bacterial interactions with the autophagic machinery (Castillo et al., 2012). Mycobacterium can activate the mTOR pathway, a key negative regulator of autophagy. By maintaining mTOR activity, Mycobacterium prevents the initiation of autophagy, hindering the formation of autophagosomes (Singh and Subbian, 2018). This is crucial because the initiation of autophagy is dependent on the inhibition of mTOR, which normally suppresses the activity of the ULK1 complex, essential for the nucleation of autophagosomes (Kim et al., 2011). Bacterium secretes proteins such as PknG, a serine/threonine kinase, which inhibits the maturation of phagosomes. PknG prevents the acidification and fusion of Mtb-containing phagosomes with lysosomes, thereby blocking their transformation into autophagolysosomes where the bacteria would be degraded (Ge et al., 2022). Mycobacterium also secretes SapM, which interacts with the adaptor protein Raptor that is involved in the mTOR pathway. SapM causes the dephosphorylation of Raptor and this interaction results in mTORC1 hyperactivity, which in turn inhibits autophagy (Zhang et al., 2024). Mycobacterial proteins PE_PGRS20 and PE_PGRS47 have been shown to interact with host autophagy proteins. These interactions can modulate autophagic flux, ensuring that the autophagy process is altered in a way that favors bacterial survival rather than its degradation. These proteins interact directly with Ras-related protein Rab1A - a multifunctional regulator in the autophagy pathway (Strong et al., 2021). Phagosomal maturation through fusion with lysosomes relies, besides others, on vacuolar ATPase, which acidifies the phagosomal lumen by hydrolyzing ATP. Mycobacterium inhibits host vacuolar ATPase using mechanisms involving the mycobacterial secreted phosphatase PtpA that interacts with vacuolar ATPase to enhance bacterial survival and pathogenicity (Wong et al., 2011; Kim et al., 2020). Two mycobacterial proteins ESAT-6 and CFP-10 are also secreted and plays a critical role in preventing phagolysosomal fusion, thereby aiding in the intracellular survival of Mycobacterium (Tan et al., 2006). Interaction of Mycobacterium with host autophagy is very well described in (Kim et al., 2020).
Listeria monocytogenes, a facultative intracellular pathogen, avoids autophagy by expressing two key determinants of pathogenesis: secreted phosphatidylinositol-specific phospholipases C (PlcA) (Mitchell et al., 2015), broad-range phospholipase C (PlcB) (Smith et al., 1995), a surface protein (ActA) (Yoshikawa et al., 2009a, Yoshikawa et al., 2009b) and pore-forming cytolysin listeriolysin O (LLO) (Schnupf and Portnoy, 2007). These factors allow the bacterium to escape from phagosomes, grow in the host cytosol, and evade the autophagic response (Mitchell et al., 2015, 2018). In addition to these proteins, the surface-associated protein InlK, encoded by the lmo1290 gene, has been identified to play a crucial role in Listeria’s ability to evade autophagy (Dortet et al., 2011). A yeast two-hybrid assay revealed that major vault protein (MVP), a highly abundant component of the eukaryotic cytoplasm, is a potential interacting partner of InlK (Dortet et al., 2011). InlK recruits MVP to coat the surface of Listeria so that the bacterium can escape autophagic recognition (Dortet et al., 2012).
In the case of Streptococcus, SpyCEP (Streptococcal pyrogenic exotoxin B cleaving enzyme) and SpeB (Streptococcal cysteine protease B) are critical factors to evade the host’s autophagic defenses, facilitating their survival and proliferation within host cells. SpyCEP is an interleukin-8 protease that is highly upregulated during invasive streptococcal infections. This protease cleaves and inactivates IL-8, a key chemokine involved in recruiting immune cells to the site of infection. By doing so, SpyCEP helps bacteria evade the immune response, including autophagy, which is a critical host defense mechanism (Bergmann et al., 2022). SpeB is a cysteine protease that is involved in the degradation of host cell proteins and immune modulators. SpeB degrades p62, NDP52, and NBR1. These proteins are essential components of the autophagy machinery, functioning as adaptors that help in recognizing and targeting bacteria for autophagic degradation. By degrading these adaptors, SpeB effectively inhibits the autophagic process within the host cell cytosol (Barnett et al., 2013).
The uptake of Yersinia by host cells triggers autophagy-related processes, but the specific pathways and outcomes vary depending on the cell type and Yersinia species. For example, Y. pseudotuberculosis has been shown to induce classical autophagy in macrophages and a variant called LC3-assisted phagocytosis in epithelial cells (Moreau et al., 2010). However, these autophagic processes do not effectively eliminate the bacteria; instead, they may support bacterial survival within host cells. The study of (Ligeon et al., 2014) found that a subset of intracellular Y. enterocolitica localizes to autophagosomal compartments within epithelial cells. Interestingly, the autophagy triggered by Y. enterocolitica did not eliminate the bacteria but rather supported their intracellular survival and multiplication. This process differed from LC3-assisted phagocytosis and resembled classical autophagy, involving core components of the autophagic machinery. The increased intracellular replication of Y. enterocolitica due to autophagy was also associated with enhanced extracellular release of the bacteria. These findings suggest that Y. enterocolitica may exploit the canonical macroautophagy pathway to promote its intracellular replication and eventual escape from infected epithelial cells (Ligeon et al., 2014; Valencia Lopez et al., 2019). Y. pestis has been shown to reside in phagosomes that acquire certain markers of late endosomes or lysosomes but do not undergo the typical acidification process. It was demonstrated that within naive macrophages, the vacuoles containing Yersinia fail to acidify. This lack of acidification is crucial for the bacteria’s survival, as it prevents the activation of lysosomal enzymes that would otherwise degrade the pathogen (Pujol et al., 2009). Yersinia produces a variety of effector proteins that play critical roles in the bacterium’s pathogenicity, particularly by disrupting host cell responses. These proteins—YopB, YopD, YopJ (known as YopP in Y. enterocolitica), YopM, YopE, YopT, YopH, YpkA (referred to as YopO in Y. enterocolitica), and YopK (YopQ in Y. enterocolitica)—interfere with the host’s immune defenses. By targeting and inhibiting key cellular processes, these Yop proteins help Yersinia survive and proliferate within the host (Seabaugh and Anderson, 2024).
It has been demonstrated that Pseudomonas aeruginosa infection also leads to the induction of autophagy (Yuan et al., 2012) but the question has arisen if P. aeruginosa, an extracellular pathogen, could modulate autophagy for its own benefit. The study of (Rao et al., 2021) has revealed that this pathogen affects the host defense pathway using T3SS. This secretion system could be used for the injection of up to four cytotoxins produced by P. aeruginosa (Hauser, 2009). The only toxin among these secreted proteins, which could dampen autophagy, is ExoS. Its mode of action is inhibition of mTOR by ADP ribosylation of Ras and concurrently inhibition of the autophagy process through repression of Vps34 kinase activity (autophagy–associated) via ADP ribosylation (Rao et al., 2021).
Bacillus anthracis produces Edema toxin (Edtx), which is a cAMP-elevating and thus capable of inhibiting autophagy as well as cholera toxin (Ctx) from Vibrio cholerae (Shahnazari et al., 2011; Huang and Brumell, 2014). Apart from Ctx, V. cholerae produces several other toxins. The V. cholerae cytolysin (VCC) protein is a key virulence factor that can disrupt host cell membranes by forming transmembrane pores, leading to cell lysis or triggering various cellular stress signaling. VCC can induce an autophagic response that leads to incomplete or stalled autophagic flux. While autophagosomes are formed in response to VCC, their maturation into autolysosomes—where degradation occurs—may be impaired, resulting in an accumulation of autophagosomes without effective breakdown of their contents (Gutierrez et al., 2007). MakA (Motility-associated killing factor A) interacts with the cellular membrane, leading to pore formation and disruption of membrane integrity. MakA is taken up by host cells, leading to the formation of cholesterol-rich membrane aggregates in a pH-dependent manner in endolysosomes, which triggers a non-canonical autophagy pathway with unconventional LC3 lipidation on these membranes (Corkery et al., 2021; Jia et al., 2022).
Enterococcus faecalis, a gram- positive opportunistic invasive bacterium and a member of human intestinum microbiota (Klare et al., 2001) has been shown to induce formation of autophagosomes in small intestinal epithelial cells (Benjamin et al., 2013). Conversely, studies by Zou and Shankar (2014) demonstrated that E. faecalis infection activates PI3K/Akt signaling pathway in host cell, potentially contributing to autophagy inhibition. Further, their research revealed that following internalization, the Enterococcus-containing vacuole (ECV) is a single-membrane organelle that resists acidification (Zou and Shankar, 2016). In contrast, Lin et al. (2018) suggested that E. faecalis lipoteichoic acid (LTA) efficiently activates macrophage autophagy. This activation is achieved by p-Akt and p-mTOR inhibition and the process is dependent on Beclin1.
Francisella tularensisFrancisella species and pathogenesisF. tularensis, a causative agent of a potentially lethal zoonotic disease tularemia, is a gram-negative facultative intracellular bacterium (Maurin, 2020). With an extremely low infectious dose (fewer than 10 colony-forming units, CFU), it is considered one of the most infectious pathogens described (Travis et al., 2021). Therefore, due to its high virulence, and multiple transmission routes with easy dissemination, the U.S. Centers for Disease Control and Prevention (CDC) classifies it as a Tier 1 Select Agent with the potential to be used as a biological weapon (Rowe and Huntley, 2015).
Currently, three subspecies are distinguished based on their metabolic characteristics, and virulence differences, such as F. tularensis, subsp. tularensis (type A), F. tularensis subsp. holarctica (type B), and F. tularensis subsp. mediasiatica. However, only F. tularensis subsp. tularensis and holarctica are known to cause tularemia in healthy individuals (Degabriel et al., 2023). While the type A strain primarily occurs on the ground in North America, the type B strain is mainly found in countries across the Northern Hemisphere (Rowe and Huntley, 2015). Although direct human-to-human transmission has not been reported, transmission through solid organ transplantation occurred in the United States in 2017, resulting in the death of one recipient (Nelson et al., 2019).
The pathogenicity of F. tularensis is primarily attributed to its ability to replicate and survive within various eukaryotic cells, especially macrophages. Infections of other cells, such as dendritic cells, hepatocytes, neutrophils, or endothelial cells, have also been documented (Bröms et al., 2010; Celli and Zahrt, 2013). During infection, the pathogen is engulfed by the macrophage through an asymmetric pseudopod loop, a process known as a,looping phagocytosis” (Clemens et al., 2004). Subsequently, the pathogen resides within a Francisella-containing vacuole (FCV), preventing phagolysosomal fusion and escaping into the nutrition-rich cytosol, where a massive replication occurs. Eventually, this process leads to cell apoptosis and infection of surrounding macrophages, thereby spreading the infection (Pechous et al., 2009; Celli and Zahrt, 2013; Ramakrishnan, 2017). The mechanisms of phagosome escape are not fully understood yet, but a gene cluster known as the Francisella pathogenicity island (FPI) has been identified as a key factor, encoding proteins essential for the constitution of the atypical type VI secretion system (T6SS) (Clemens et al., 2015; Rigard et al., 2016; Spidlova and Stulik, 2017). Interestingly, none of those proteins possess properties of cytolysins, pore-forming toxins, or hydrolytic enzymes, suggesting a novel bacterial escape mechanism (Bröms et al., 2010). In addition to FPI proteins, F. tularensis virulence is critically dependent on several other factors, including MglA, SspA, PigR (also known as FevR), ppGpp, (Lauriano et al., 2004; Wrench et al., 2013) and the HU protein (Stojkova et al., 2018, Stojkova et al., 2019; Stojkova and Spidlova, 2022). These proteins, along with various other transcription factors (Spidlova et al., 2020), play essential roles in regulating virulence gene expression and ensuring the pathogen’s ability to survive and proliferate within the host. Their coordinated action is vital for the bacterium’s pathogenicity and ability to evade host immune responses.
Molecular insights into Francisella-autophagy interactionThe role of autophagy in the host defense against members of the Francisella genus is controversial (Qi et al., 2016). Comparative studies indicate that the less virulent LVS genome has undergone significant rearrangements compared to fully virulent SchuS4 strain. These rearrangements include inversions and deletions, leading to differences in gene content and organization. For instance, certain genes present in SchuS4 are either absent or pseudogenized in LVS, potentially affecting pathogenicity (Chaudhuri et al., 2007). While both strains possess the FPI, variations in gene sequences and expression levels have been observed, which may account for differences in their virulence (Faron et al., 2013). The distinct immune response dynamics observed between SchuS4 and LVS strains highlight differential regulation of key cellular processes, including the induction or suppression of autophagy, which may significantly impact their pathogenic strategies and host interactions. At an early stage of infection, F. tularensis dampens the autophagy process. The reasons why are still unanswered but a few possible explanations exist: a) F. tularensis prefers replication in cytosol instead of phagosome, b) the delay could bring time to become resistant to the autophagolysosome’s acidic environment. On the other hand, at late stages of infection, F. tularensis exploits autophagy to be hidden inside autophagosomes, which leads to suppression of proinflammatory cytokines production (Cremer et al., 2009). F. tularensis avoids autophagic degradation by escaping from the phagosome before it can be targeted by autophagic machinery. After being phagocytosed by host cells, F. tularensis rapidly escapes into the cytosol, thereby avoiding the lysosomal degradation pathway (Checroun et al., 2006). The bacterial factors that mediate this escape are crucial for avoiding recognition by the autophagy machinery. The study has shown that the IglC protein is essential for phagosomal escape and subsequent replication in the cytosol (Santic et al., 2005). PdpC and PdpD that were identified as T6SS effectors (Eshraghi et al., 2016) are required for phagosomal escape (Ludu et al., 2008; Uda et al., 2016) and OpiA, a phosphatidylinositol 3-kinase that is not encoded in FPI, is responsible for delaying phagosomal maturation (Ledvina et al., 2018). These effector proteins contribute to Francisella virulence (Brodmann et al., 2021). Similarly, many other studies have described various proteins that are necessary for the intracellular replication of F. tularensis inside the host cell (Bröms et al., 2010; Barel and Charbit, 2013; Celli and Zahrt, 2013; Ozanic et al., 2016; Alam et al., 2018; Spidlova et al., 2018; Stojkova et al., 2018). After F. tularensis enters the host cell (usually a macrophage), it is initially enclosed in a membrane-bound compartment known as the Francisella-containing vacuole. This is a phagosome-like structure formed during the phagocytosis process. Once F. tularensis escapes the FCV and replicates in the cytosol, the host cell may attempt to target the bacterium for destruction through autophagy, a process where cellular debris or pathogens are engulfed by autophagosomes (double-membrane structures) and delivered to lysosomes for degradation. F. tularensis interferes with the autophagy machinery by negative modulating the expression of autophagy-related genes and proteins, including BECN1, ATG5, ATG12, ATG16L2, ATG7, and ATG4a (Cremer et al., 2009). It is known that there exist two type of autophagy process: a) ATG5- dependent autophagy and b) ATG5-independent autophagy. In order to increase intracellular stocks of host amino acids, which may be utilized as a source of carbon, energy, and iron, F. tularensis induces ATG5-independent autophagy (Steele et al., 2013). It has been shown that WT strains of Francisella species are able to resist ATG5-dependent autophagy (Chong et al., 2012; Case et al., 2014). But it seems that the WT strains somehow exploit ATG5-dependent autophagy or responses to this pathway, as shown in an example of LVS. This strain proliferates less effectively in ATG5-deficient mice when compared to the WT mice (Kelava et al., 2020). Contrarily the mutant strains unable to replicate within the host or deficient in O-antigen synthesis are captured by ATG5-dependent autophagy (Case et al., 2014). Highly virulent strain F. tularensis subsp. tularensis SchuS4, which successfully avoids being recognized by the autophagic machinery, does not undergo ubiquitination (a critical step for autophagic targeting) in the cytosol and SchuS4 bacteria are not fully recognized by the key autophagy receptors p62/SQSTM1 and NBR1 (Chong et al., 2012), compared to the F. tularensis subsp. holarctica LVS that induces the recruitment of p62/SQSTM1 and LC3 already after 1 hour post infection (Härtlova et al., 2014). On the other hand, SchuS4 mutants that are not able to survive in the cytosol are tagged with ubiquitin and are subsequently captured into autophagosomes in a process dependent on ATG5, LC3, and p62/SQSTM1 (Chong et al., 2012). Tagging of bacteria by ubiquitin is a critical step for recognition in autophagic process and many bacteria can manipulate with this ubiquitination/deubiquitination system for their benefit (Vozandychova et al., 2021) and likewise F. tularensis that is able to suppress the activity of deubiquitinating enzymes and thus disrupt the homeostasis in ubiquitin cycle (Vozandychova et al., 2023). F. tularensis subsp. holarctica FSC200 downregulates the activity of USP10 enzyme in human macrophages 1 hour post infection, leading to the decreased amount/degradation of LC3, and thus repression of autophagy. USP10 normally removes ubiquitin molecule from the Beclin1 and LC3 (required for autophagosome formation) and thus these proteins are not degraded (because they are not tagged by ubiquitin). This suggests an active manipulation of the autophagy by F. tularensis specific strain (Vozandychova et al., 2023). Francisella’s HU protein (Stojkova et al., 2019), a DNA-binding protein involved in pathogenesis and virulence (Stojkova et al., 2018), may play a regulatory role in host´s response. It has already been shown in other pathogens that the bacterial HU protein is capable of binding host DNA (Stojkova and Spidlova, 2022). Since it has been demonstrated that F. tularensis HU protein is secreted into the medium (Konecna et al., 2010), we can speculate whether it enters the host cell, or even host nucleus where it could bind host DNA, because the F. tularensis HU protein´s DNA binding motif that we identified in our previous study (Pavlik and Spidlova, 2022) can be found in the host genome (Genome Data Viewer, NCBI). By binding to the promoter regions or regulatory elements of these genes, HU protein could affect their transcription, thus modulating the expression of key components in the autophagy process.
The key question remains regarding the dual role of autophagy as both a defense mechanism and a resource exploited by Francisella species. For instance, the exploitation of autophagy by F. tularensis at different infection stages, such as its ability to evade recognition by suppressing ubiquitination or manipulating ATG5-independent pathways, represents not just a mechanistic insight but a potential focal point for therapeutic intervention. So far, we know only a few of bacterial effector proteins that are somehow included in affecting the process of autophagy and their exact mechanisms of action remain elusive. It is crucial to delve deeper into the molecular interactions between bacterial effector proteins and host proteins that regulate autophagy machinery. Uncovering these detailed mechanisms can guide future research toward the development of targeted inhibitors, offering new strategies to combat infections.
ConclusionThe study of Francisella provides general insight into the fight against intracellular bacterial pathogens, as many of these organisms share similar strategies for avoiding autophagy. Understanding the molecular interplay between host autophagy and microbial evasion tactics may be the basis for the development of treatments not only against tularemia but also against a number of other infectious diseases. Additionally, the insights gained from F. tularensis research have the potential to extend beyond infectious diseases and offer new approaches to manipulate autophagy for therapeutic benefit in cancer, autoimmune diseases, and other conditions where autophagy plays a critical role. Once the molecular interactions between F. tularensis and the host autophagy machinery are better understood, novel regulators of autophagy may be identified. These could include host proteins that are modulated by F. tularensis to suppress autophagy or new bacterial factors that inhibit autophagic processes. Future research should focus on uncovering the molecular details of Francisella-host interactions, characterizing the role of host genetic factors in autophagy response, and developing novel drugs and vaccine strategies that can modulate autophagy.
Author contributionsPP: Conceptualization, Validation, Visualization, Writing – original draft, Writing – review & editing. EV: Writing – original draft, Writing – review & editing. PS: Conceptualization, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The work was supported by the Ministry of Defence of the Czech Republic “Long Term Organization Development Plan 1011” – Healthcare Challenges of WMD II of the Military Faculty of Medicine Hradec Kralove, University of Defence, Czech Republic (Project No: DZRO-FVZ22-ZHN II).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ReferencesAbd El Maksoud, A. I., Elebeedy, D., Abass, N. H., Awad, A. M., Nasr, G. M., Roshdy, T., et al. (2019). Methylomic changes of autophagy-related genes by legionella effector lpg2936 in infected macrophages. Front. Cell Dev. Biol. 7. doi: 10.3389/fcell.2019.00390
PubMed Abstract | Crossref Full Text | Google Scholar
Alam, A., Golovliov, I., Javed, E., Sjöstedt, A. (2018). ClpB mutants of Francisella tularensis subspecies holarctica and tularensis are defective for type VI secretion and intracellular replication. Sci. Rep. 8, 11324. doi: 10.1038/s41598-018-29745-4
PubMed Abstract | Crossref Full Text | Google Scholar
Arasaki, K., Mikami, Y., Shames, S. R., Inoue, H., Wakana, Y., Tagaya, M. (2017). Legionella effector Lpg1137 shuts down ER-mitochondria communication through cleavage of syntaxin 17. Nat. Commun. 8, 15406. doi: 10.1038/ncomms15406
PubMed Abstract | Crossref Full Text | Google Scholar
Ashida, H., Mimuro, H., Sasakawa, C. (2015). Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6. doi: 10.3389/fimmu.2015.00219
PubMed Abstract | Crossref Full Text | Google Scholar
Barnett, T. C., Liebl, D., Seymour, L. M., Gillen, C. M., Lim, J. Y., LaRock, C. N., et al. (2013). The globally disseminated M1T1 clone of Group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14, 675–682. doi: 10.1016/j.chom.2013.11.003
PubMed Abstract | Crossref Full Text | Google Scholar
Baxt, L. A., Goldberg, M. B. (2014). Host and bacterial proteins that repress recruitment of LC3 to shigella early during infection. PLoS One 9, e94653. doi: 10.1371/journal.pone.0094653
PubMed Abstract | Crossref Full Text | Google Scholar
Beck, W. H. J., Kim, D., Das, J., Yu, H., Smolka, M. B., Mao, Y. (2020). Glucosylation by the legionella effector setA promotes the nuclear localization of the transcription factor TFEB. iScience 23, 101300. doi: 10.1016/j.isci.2020.101300
PubMed Abstract | Crossref Full Text | Google Scholar
Benjamin, J. L., Sumpter, R., Levine, B., Hooper, L. V. (2013). Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13, 723–734. doi: 10.1016/j.chom.2013.05.004
PubMed Abstract | Crossref Full Text | Google Scholar
Bergmann, R., Gulotta, G., Andreoni, F., Sumitomo, T., Kawabata, S., Zinkernagel, A. S., et al. (2022). The group A Streptococcus interleukin-8 protease SpyCEP promotes bacterial intracellular survival by evasion of autophagy. Infect. Microbes Dis. 4, 116–123. doi: 10.1097/im9.0000000000000098
PubMed Abstract | Crossref Full Text | Google Scholar
Bhogaraju, S., Kalayil, S., Liu, Y., Bonn, F., Colby, T., Matic, I., et al. (2016). Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 167, 1636–1649.e13. doi: 10.1016/j.cell.2016.11.019
PubMed Abstract | Crossref Full Text | Google Scholar
Brodmann, M., Schnider, S. T., Basler, M. (2021). Type VI secretion system and its effectors pdpC, pdpD, and opiA contribute to Francisella virulence in galleria mellonella larvae. Infection Immun. 89, e0057920. doi: 10.1128/iai.00579-20
PubMed Abstract | Crossref Full Text | Google Scholar
Bröms, J. E., Sjöstedt, A., Lavander, M. (2010). The role of the Francisella tularensis pathogenicity island in type VI secretion, intracellular survival, and modulation of host cell signaling. Front. Microbiol. 1. doi: 10.3389/fmicb.2010.00136
PubMed Abstract | Crossref Full Text | Google Scholar
Case, E. D. R., Chong, A., Wehrly, T. D., Hansen, B., Child, R., Hwang, S., et al. (2014). The rancisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell. Microbiol. 16, 862–877. doi: 10.1111/cmi.12246
PubMed Abstract | Crossref Full Text | Google Scholar
Castillo, E. F., Dekonenko, A., Arko-Mensah, J., Mandell, M. A., Dupont, N., Jiang, S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. U. S. A. 109, E3168–E3176. doi: 10.1073/pnas.1210500109
PubMed Abstract | Crossref Full Text | Google Scholar
Celli, J. (2019). The intracellular life cycle of brucella spp. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.bai-0006–2019
Crossref Full Text | Google Scholar
Chatterjee, R., Chaudhuri, D., Setty, S. R. G., Chakravortty, D. (2023). Deceiving the big eaters: Salmonella Typhimurium SopB subverts host cell xenophagy in macrophages via dual mechanisms. Microbes Infection 25, 105128. doi: 10.1016/j.micinf.2023.105128
PubMed Abstract | Crossref Full Text | Google Scholar
Chaudhary, A., Kamischke, C., Leite, M., Altura, M. A., Kinman, L., Kulasekara, H., et al. (2018). [amp]]beta;-Barrel outer membrane proteins suppress mTORC2 activation and induce autophagic responses. Sci. Signaling 11, eaat7493. doi: 10.1126/scisignal.aat7493
PubMed Abstract | Crossref Full Text | Google Scholar
Chaudhuri, R. R., Ren, C.-P., Desmond, L., Vincent, G. A., Silman, N. J., Brehm, J. K., et al. (2007). Genome sequencing shows that European isolates of Francisella tularensis subspecies tularensis are almost identical to US laboratory strain Schu S4. PLoS One 2, e352. doi: 10.1371/journal.pone.0000352
PubMed Abstract | Crossref Full Text | Google Scholar
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., Celli, J. (2006).
留言 (0)