
記住我
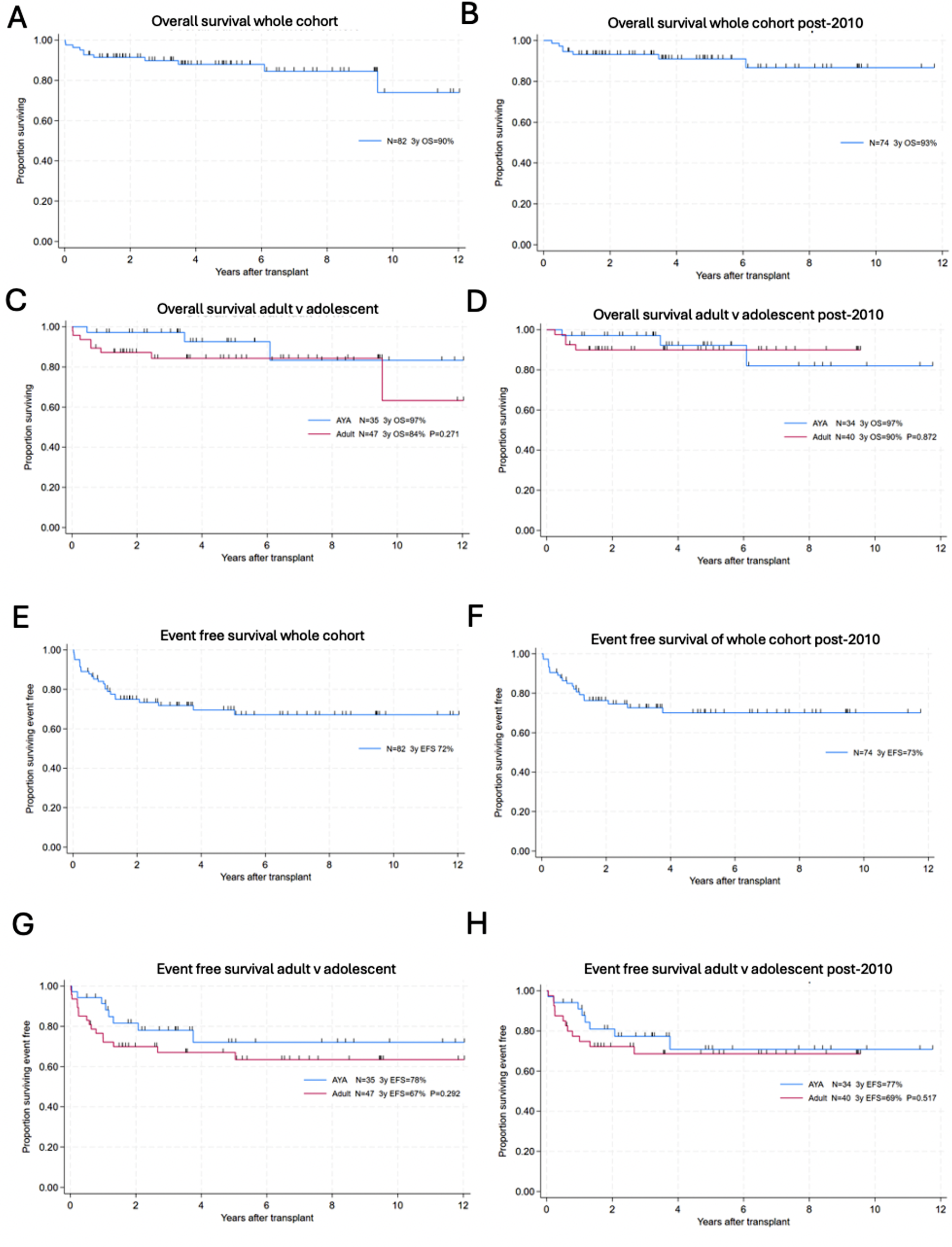
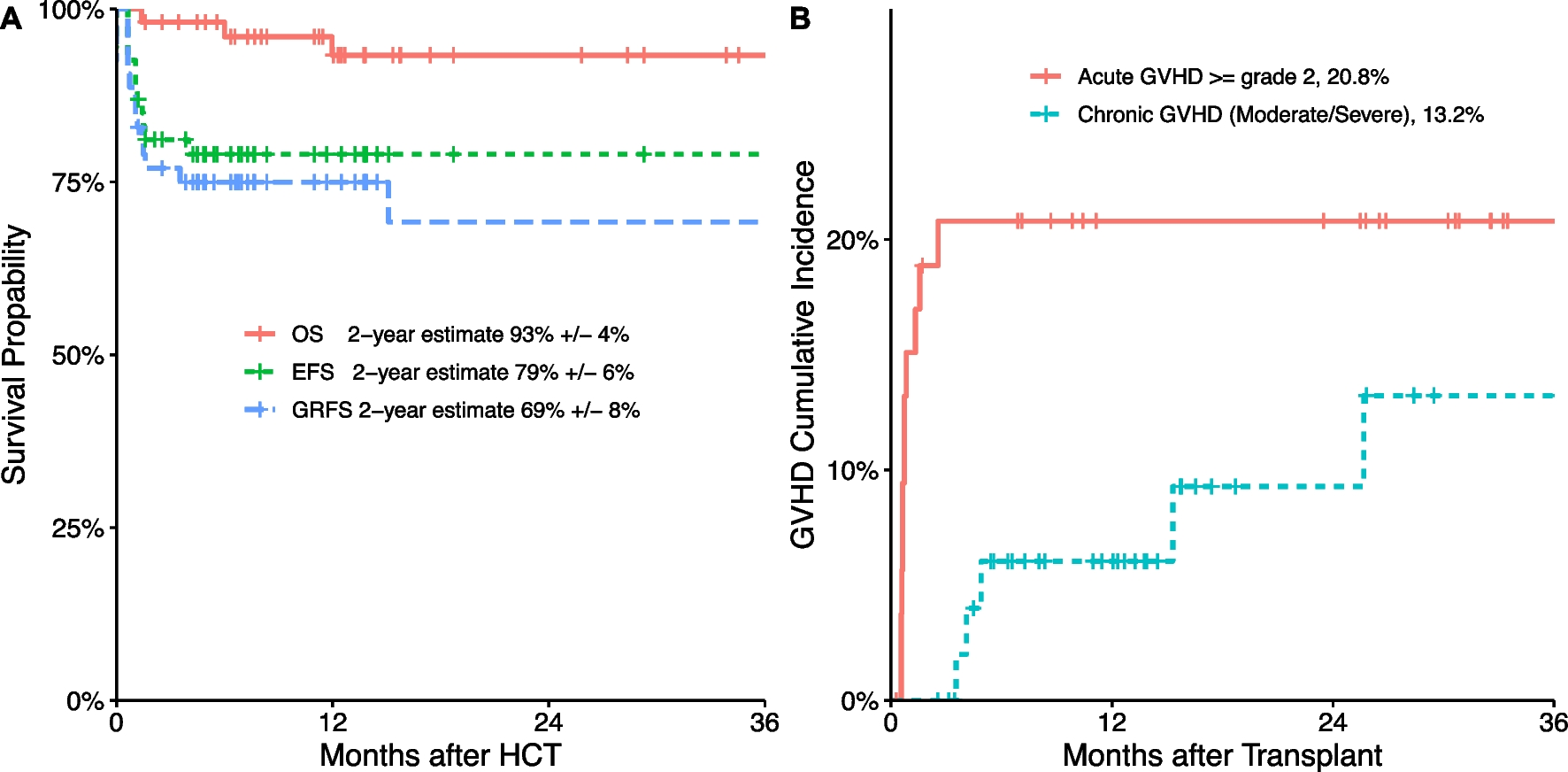


Twenty-six patients were alive at the time of data collection, and had a median age of 19.9 years (3.6–57.5 years). The median follow-up time was 74 months (6–384 months).Two patients died, at the age of 49: p13 due to SARS-CoV-2 infection and non-Hodgkin lymphoma (NHL); p27 following a heart attack, not related to the underlying disease.
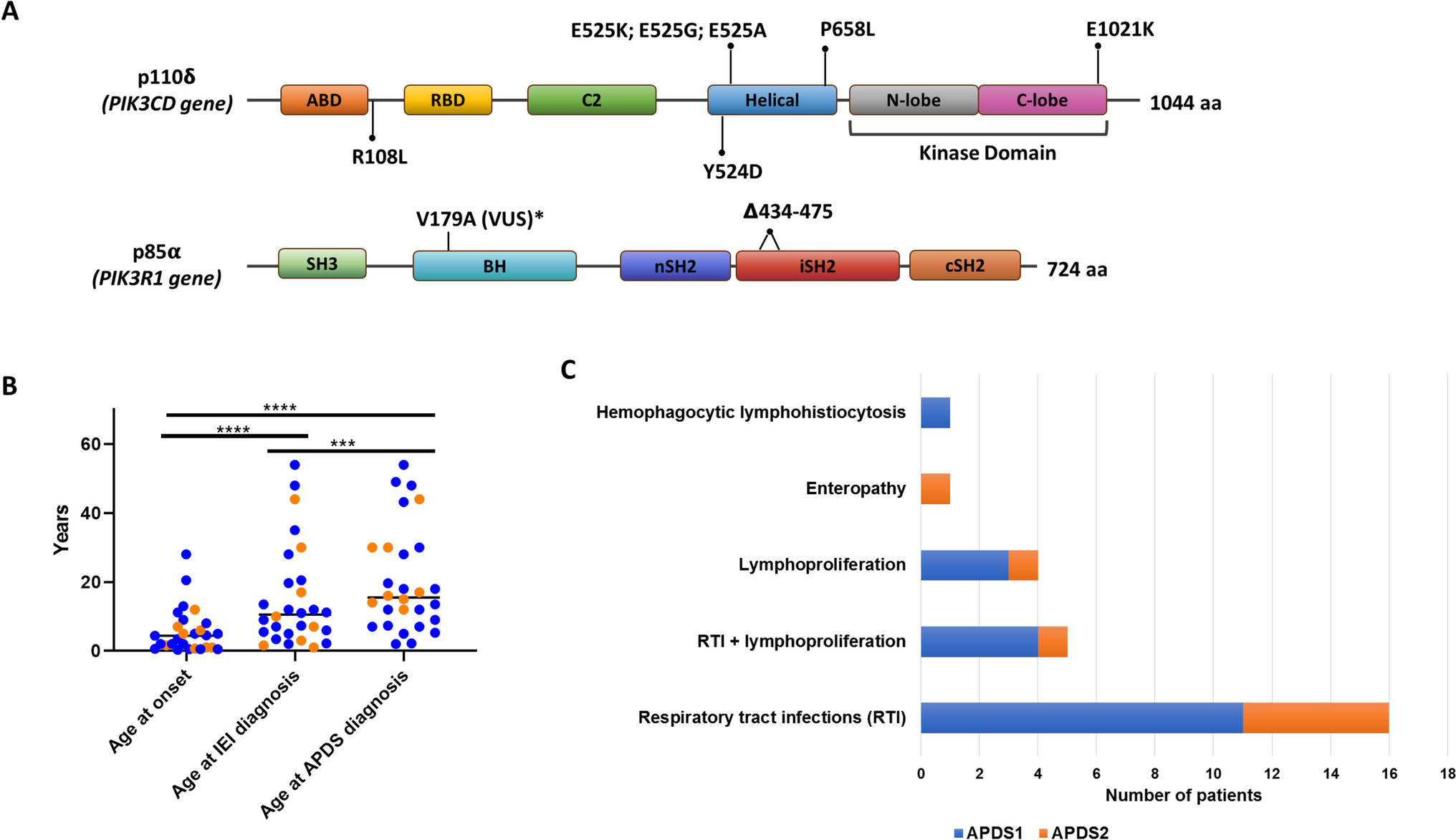
GeneticsTwenty out of 28 APDS patients analyzed have been genetically diagnosed with APDS1. Eleven patients carried the heterozygous E1021K mutation, and one patient had the E525A mutation in the PIK3CD gene. The mutations Y524D, R108L, and P658L have been proven pathogenic by S6 phosphorylation assay [14]. P4 carried the E525G mutation in PIK3CD, pathogenicity was confirmed by hyperphosphorylation in the S473 residue of AKT [24]. Of note, p13 had both the pathogenic mutation E525K in PIK3CD and an additional heterozygous rare VUS in PIK3R1 (V179A).
All 8 patients diagnosed with APDS2 presented splicing site mutations, either in position c.1300–2 A > G or c.1425 + 1 G > C. Independently of the position or the nucleotide change, both variants led to the deletion of 41 amino acids in the inter-SH2 domain. The distribution of the variants across the protein domains is represented in Fig. 1A.
Fig. 1
Clinical and molecular diagnosis of APDS cohort: gene variants (A), diagnostic delay (B) and clinical signs at onset (C). Abbreviations: ABD, Adaptor-binding domain; BH, Bcl-2 homology domain; cSH2, C-terminal SH2; nSH2, N-terminal SH2; RBD, Ras-binding domain; SH, Src homology domain; VUS, Variant of Uncertain Significance; APDS, Activated PI3K-kinase Delta Syndrome; IEI, Inborn Errors of Immunity
Table S1 provides a summary of the patient’s genetic diagnosis and references for published validation assays.
Clinical Characteristics Clinical Features at OnsetThe median age of the clinical presentation of APDS was 4.4 years (3 months − 28 years), while the diagnosis of IEI arose only later in the evolution of the disease (median age 10.5 years; 1–54 years). Finally the patients reached the APDS diagnosis at a median age of 15.5 years (2–54 years) (Fig. 1B).
The main presenting symptoms were RTI alone (16/28) or associated with lymphoproliferation (5/28). Four patients presented isolated lymphoproliferation, non-clonal at the onset. Other presenting signs were enteropathy and one case of autoinflammatory symptoms with a hemophagocytic lymphohistiocytosis-like picture (Fig. 1C).
Two patients experienced adult onset: p19 had recurrent RTI from the age of 20.5; p15, at 28 years, experienced lymphoproliferation (adenopathies, tonsil hypertrophy, and hepatosplenomegaly).
One patient, diagnosed due to affected family members carrying the pathogenic mutation p.Y524D, is still asymptomatic at the age of 57 years.
InfectionsOver time, up to 78.6% of patients (n = 22) presented recurrent RTI despite their absence at onset. Moreover, 57% of patients needed prolonged antibiotic therapies (> 2 weeks) suggesting severe infections.
Six patients had other infections (severe gastroenteritis, sepsis, impetigo). One patient (p22) with a CID (combined immunodeficiency) phenotype resulted positive for atypical mycobacteria on bronchoalveolar lavage; he was treated for eleven months and relapsed seven months after the therapy withdrawal.
Twelve patients (42.9%) had chronic positive EBV viremia in peripheral blood, while EBV-related symptoms were detected in only 9 patients (32% of the cohort). All the symptomatic patients had chronic viremia. Four out of six patients presenting chronic positive EBV viremia were tested for EBV serology and showed persistent positivity of IgM against the virus capsid antigen (VCA), while IgG serology was not informative due to immunoglobulin supplementation. Chronic positive CMV viremia was less common (6/28), mildly symptomatic only in one case. Other patients presented with fever and splenomegaly but completely cleared the virus after antiviral therapy.
Non-malignant LymphoproliferationSystemic non-clonal lymphoproliferative disorders (SNCLD) characterized 89% of patients (25/28). At a median age of 7 years (range 0.6–30), they showed multiple chronic lymphadenopathies, thoracic or gastrointestinal tract lymphoid hyperplasia, granulomatous lymphocytic interstitial lung disease (GLILD), tonsil hypertrophy, splenomegaly, and hepatomegaly.
Splenomegaly, affecting 22/28 patients, was the leading expression of SNCLD and persisted for more than six months in 19/22 of cases, associated with hepatomegaly in 8/28 individuals. Nineteen patients showed chronic lymphadenopathies, combined with thoracic and gastrointestinal lymphoid hyperplasia. Uncommon manifestations were tonsil hypertrophy (8/28) and GLILD (3/28).
Malignant Lymphoproliferation and Other MalignanciesClonal lymphoproliferative disorders occurred in 4 (14%) patients at a median age of 18 years (17–19). Two patients experienced gastrointestinal EBV-positive diffuse large B cell lymphoma (DLBCL), at the age of 19 (p22) and 18 years (p24), respectively. In the former case, a duodenal DLBCL was followed by a further caecal lymphoma with histologic features of Hodgkin lymphoma (HL)/DLCBL; in the latter, a colonic DLBCL was preceded and followed by a cervical nodular lymphocyte-predominant HL. Furthermore, there was a case of EBV-positive stage IV anaplastic lymphoma in a 17-year-old patient (p27) and a case of EBV-positive NHL in a 47-year-old patient (p13) (Fig. 2). All patients required multiple-line chemotherapies to induce remission. Nonetheless, p13 died at 49 years following SARS-CoV-2 infection and NHL relapse. Further data about malignant lymphoproliferation are reported in Tables 1 and 2.
Fig. 2
Malignant lymphoproliferation in the APDS cohort. Abbreviations: APDS, Activated PI3K-kinase Delta Syndrome; AL, Anaplastic lymphoma; DLBCL, Diffuse Large B Cell Lymphoma; EBER, EBV-encoded RNA; EBV, Epstein-Barr virus; HL, Hodgkin Lymphoma; NHL, Non-Hodgkin Lymphoma; NLPHL, Nodular lymphocyte-predominant Hodgkin Lymphoma. Adapted from Rivalta B et al. Case Report: EBV Chronic Infection and Lymphoproliferation in Four APDS Patients: The Challenge of Proper Characterization, Therapy, and Follow-Up. Front Pediatr. 2021 Aug 27;9:703853. https://doi.org/10.3389/fped.2021.703853. PMID: 34540765; PMCID: PMC8448282
Table 1 Clinical features of the APDS cohortTable 2 Detailed features of the malignant lymphoproliferative manifestations in the cohortOf note, patient (p9) was initially diagnosed at the age of 20 with HL requiring multiple therapies: six-cycle ABVD, two-cycle IEV, and autologous HSCT. Fourteen years later, the first lymph node biopsy was reviewed, recanting the previous HL diagnosis in favor of non-malignant hyperplastic lymphadenopathy with relevant activation of the interfollicular area by CD30+, mostly mononucleated cells. This was superimposable to a follow-up biopsy performed ten years later, after therapies.
The only other malignancy was an ovarian dysgerminoma occurring in a 14-year-old female (p7).
Lung ComplicationsNon-infectious lung complications affected 54% of patients, aged between 5 and 40 years (median 10 years). CT scans identified bronchiectasis, nodules, and GLILD in 43%, 25%, and 11% of patients, respectively. Additional lung complications were thoracic lymphadenopathies (3 cases) and partial lung dystelectasis with aspecific inflammatory reaction (1 patient).
Gastrointestinal Benign ManifestationsGastrointestinal benign disorders involved 43% of patients (7/20 APDS1 vs. 5/8 APDS2), at a median age of 18.5 years (range 2–45). Intestinal lymphoid hyperplasia and inflammatory colitis were the most frequent, affecting 4/28 individuals, the latter associated in 2 cases with an intestinal DLBCL. Other gastrointestinal non-neoplastic disorders are specified in Table 1.
Autoimmune DisordersAutoimmune manifestations occurred in the 21% (6/28) of patients, with a median onset age of 19.5 years (5–42 years). The leading manifestation was autoimmune hemolytic anemia (AIHA), affecting 4/28 patients, combined in one case with autoimmune hepatitis (p22), in another with a systemic lupus erythematosus (SLE)-like condition (p9), followed by immune thrombocytopenia with renal disease (C3-glomerulopathy in p11), and thyroiditis with inflammatory bowel disease (IBD) (p24).
A quarter of patients presented cytopenia, with anemia being the most common (21%; n = 6), followed by thrombocytopenia (4 cases) and neutropenia (1 case). Immune cytopenias were more frequent in APDS2 than APDS1 patients (4/8 vs. 2/20). Multiple causes of cytopenia, such as autoimmunity, concomitant infection, or chronic blood loss, could be recognized; often contemporary present in a single patient .
Neurological DisordersNine patients (32%) displayed some neurological manifestations, most commonly identified at a median age of 6.7 years (range 3–31). Mild cognitive impairment was reported in 7 patients by physicians during clinical evaluation, although it was not confirmed by intellectual quotient evaluation in the majority of cases. Associated features were anxiety and facial paresthesia, epilepsy, and posterior reversible encephalopathy syndrome (the latter probably related to kidney disease).
Immunological ProfileThe immunological assessment of this patients’ cohort reveals a wide range of cellular and humoral abnormalities (Table 3). Eight out of 28 (29%) showed low absolute counts of T cells. Expansion of senescent CD57 + CD3 + T cells or effector memory subsets was the most common T cell alteration, detected in 54% of patients, usually associated with normal T cell counts, except for 1 patient with T cell lymphopenia. Similarly, 32% of patients showed a reduction of naïve T cell (CD4 + or CD8+). Double-negative αβ T cells and γδ T cells were increased in a minority of cases (2 and 1 out of 28, respectively).
Table 3 Immunological features of the APDS cohortB cells lymphopenia occurred in 43% of cases. Independently from the total B cell count, 54% of patients displayed low switched memory B cells (in the majority of cases coupled with an abnormal T cell phenotype). Moreover, 36% of patients presented high transitional B cell counts. In the B cell compartment, less common features were a reduction of naïve B cells and increase of plasmablasts (11% each).
In line with the B cell phenotype observed, humoral abnormalities were frequent: high IgM levels (39%), low IgA levels (39%), low IgG levels (21%), while elevated IgG and IgA levels were less frequent.
As illustrated in Table 3, most patients showed variable combinations of T and B cells defects (20/28, 71%). Few cases presented with immunological profiles resembling other primary immunodeficiencies, such as: combined immunodeficiency (4/28), autoimmune lymphoproliferative syndrome (1/28), common variable immunodeficiency (1/28), hyper-IgM syndrome (1/28). This heterogeneous immunological profile highlights both T and B cell dysregulation as hallmark features of APDS, particularly affecting memory and naïve B cell populations, as well as senescent T cell expansion in over half of the patients.
Treatment Supportive/Symptomatic TreatmentMost patients (n = 19) received Ig supplementation, all of them suffered from recurrent RTI. Eight out of 19 had hypogammaglobulinemia. The remaining 11 patients received Ig supplementation despite normal to high IgG serum levels: 7/11 due to lack of specific antibody response; 3/11 due to RTI and considering the molecular diagnosis of APDS; one patient started supplementation just for clinical indication due to RTI and benefited.
Thirteen patients underwent additional prophylaxis with either trimethoprim/sulfamethoxazole or azithromycin. One patient received azithromycin without immunoglobulins due to recurrent RTI and bronchiectasis.
Compared to the onset of infectious episodes, prophylaxis was introduced later. Median delay time was: 5.5 years for Ig supplementation (range 0.1–33); 6 years for antimicrobial prophylaxis (range 0–39).
SteroidsOnly 5/28 patients received steroid immunosuppressive therapy starting at a median age of 21 years (range 3–40). None of the patients showed a complete and persistent clinical benefit from steroids alone. Two patients needed multiple immunosuppressants to partially control immune dysregulation (SLE and IBD, respectively) (Table 2S).
PI3K/Akt/mTOR Inhibitor TreatmentRapamycin, an inhibitor of the mammalian target of rapamycin (mTOR), can modulate the dysregulated PI3Kδ-signaling pathway [33, 34], reducing lymphoproliferation [34, 35] and improving immune dysregulation even in the long-term [6]. Rapamycin was introduced at a median age of 14 (3–31 years) in 10/28 patients (36%). Two patients received rapamycin after other immunosuppressants, in 8 patients it was first-line therapy. Eight (80%) patients showed remarkable improvement of non-clonal lymphoproliferation, benefit was absent or partial in only two cases. Overall, rapamycin was well tolerated without any significant side effects. P28 showed partial improvement with theophylline administration, which seems to inhibit mTOR pathway [26] (Table 2S).
Selective PI3Kδ InhibitorsSelective PI3Kδ inhibitors are a promising therapeutic approach for APDS. In Italy, leniolisib is only available for Compassionate Drug Use (CDU).
Nine patients (32%) accessed the selective PI3Kδ inhibitor leniolisib as tailored therapy with a high safety profile and improvements: reduction of infection rate, lymphoproliferation, lung complications and autoimmunity (Fig. 3). The median follow-up under therapy is 4.5 years (range 0.2–5). Retrospective data collection does not allow a comprehensive analysis, however, for 4 patients more detailed information are available. Patient p6 did not show EBV reactivation or thrombocytopenia relapses, and a significant reduction of the spleen and liver size was documented; p9 did not have a recurrence of infections or cytopenias and improved erythema pernio as well as lymphocyte typing; p26 normalized transitional and naive B cells and reduced hyper-IgM; p28 normalized naive B cells and slightly increased memory unswitched and switched B cells (Table 3S). The first clinical trial on selective PI3Kδ inhibitors efficacy was conducted in 2015–2016 on patients older than 12: the median age at starting treatment in our cohort was 22 years (range 12–33 years). Before receiving leniolisib, p25 and p28 took part in phase 1b, an open-label, multicenter, exploratory study aimed to assess the safety, tolerability, pharmacokinetics, and efficacy of seletalisib (2015–002900-10) and its extension study (2015–005541). Even with this compound, patients improved clinically with favorable risk-benefit profiles [9]. Patients p6, p14, p18, p26, and (after the end of seletalisib clinical trial) p25, participated in a randomized, placebo-controlled phase 3 trial on leniolisib (#NCT02435173) in 2015–2022 and its extension study (#NCT02859727) currently ongoing (Table 2S) [32, 36].
Fig. 3
Clinical phenotype of APDS patients receiving leniolisib prior to treatment and after initiation. The graph represent the variation of clinical signs before (dark grey) and after (light grey) starting leniolisib
Hematopoietic Stem Cell TransplantationThree patients (p1, p2, p8) affected by APDS1 underwent hematopoietic stem cell transplantation (HSCT) at 9.9, 7.3, and 14.5 years of age. Indications to transplant were either recurrent infections despite ongoing prophylaxis (p1) and lymphoproliferation poorly responsive to rapamycin (p2, p8). Donors were all unrelated (9/10-HLA matched for p1 and p2; 10/10 for p8). All patients reached complete remission of the disease manifestations. Graft versus Host Disease (GVHD) was observed only in p1, experiencing grade III intestinal aGVHD at day + 21, grade II skin aGVHD at day + 30. She developed an autoimmune neurological complication 18 months after HSCT with central nervous system lesions detectable at magnetic resonance, requiring prolonged immunosuppression. Immunosuppressive drugs were progressively tapered and withdrawn except for immunoglobulins, continued for immune-modulatory purposes, without relapse. None of the other two patients is currently under immunosuppression or anti-infectious prophylaxis. Median follow-up after HSCT was 4 years at the time of data collection. All patients are currently alive and well, maintaining full donor chimerism (Table 4S).
留言 (0)