Approvals
Protocols for mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Penn. Patients were enrolled in an institutional review board (IRB)-approved longitudinal clinical study (REVEAL study) at Penn.
Mice
Human LSL TREX1 V235Gfs and LSL WT TREX1 transgenic mice were generated by the Miner laboratory at Penn and were previously described [5]. These animals have a CAG promoter and lox-stop-lop sequence preceding either the human WT TREX1 cDNA or TREX1 V235Gfs cDNA in the ROSA26 locus. Transgenic human TREX1 animals were crossed to tamoxifen-inducible CAG-Cre transgenic mice (Jax 004682). Mice were housed in pathogen-free facilities at Penn and fed a standard diet ad libitum. Experiments were performed using mice of both sexes at ages 8–12 weeks.
Cell Culture Studies of TREX1
Of note, the TREX1 V235Gfs mutant is toxic to cells, requiring flow cytometric screening to confirm expression. This is an absolute requirement for proper studies of RVCL-causing TREX1 variants, since only a small percentage of cells can tolerate expression of the RVCL-causing mutant.
For in vitro development of the gene therapy and localization studies, we utilized 293T cells cultured in Dulbecco’s modified eagle medium (DMEM; Gibco, 11995081) supplemented with 10% fetal bovine serum (FBS), 2 mM L-GlutaMAX (Gibco, 35050061), 1X non-essential amino acids (Gibco, 11140050), 1 mM sodium pyruvate (Gibco, 11360070), 10 mM HEPES (Gibco, 15630080), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, 15140122) at 37˚C and 5% CO2. To examine protein expression and to develop gene therapies, the WT TREX1 and TREX1 V235Gfs cDNAs were synthesized by Genscript, (Piscataway, NJ), including an N-terminal HA tag, and cloned into the multiple cloning site of the doxycycline-inducible pTRIPZ vector (AgeI/MluI). To produce pTRIPZ-TREX1 E266X, pTRIPZ-WT TREX1 was mutagenized via PCR with the following primers: 5’ GCCTTGGATAGAGCAGGGGTACCA 3' and 5’ TGGTACCCCTGCTCTATCCAAGGC 3'. Linear PCR amplicons were treated with DpnI for 3 hours at 37 °C and used to transform XL1-Blue chemically competent cells. The sequences of all plasmids were confirmed using Sanger sequencing. To produce lentivirus, 293T cells were seeded at a density of 5 × 105 cells per well in a 6-well plate and were transfected with a 4:2:1 ratio of pTRIPZ: psPAX2:VSVG for a total DNA mass of 2.5 µg/well. Cells were incubated for 72 hours. After incubation, supernatants were filtered through 0.45 μm syringe filters, aliquoted and stored at -80 °C.
To create a stable cell line with inducible expression of WT TREX1 or TREX1 E266X, 293T cells were seeded at a density of 5 × 105 cells per well in the wells of a 6 well plate. pTRIPZ-TREX1 lentiviral supernatants were diluted 1:5 in DMEM supplemented with 8 µg/mL polybrene and incubated for 72 hours. Following incubation, cells were reseeded in 10 cm dishes in DMEM supplemented with 1.5 µg/mL puromycin and cultured for 7 days. TREX1 expression was induced by culturing cells in DMEM with doxycycline (500 ng/mL) for 2 days. Longer periods of TREX1 induction induces selection events in cells expressing the RVCL-causing TREX1 variants, since these mutants are now well-known to cause DNA damage in cultured cells and in animals.
SDS PAGE and Western Blotting
Liver samples were homogenized in RIPA buffer (CST, 9806) with protease inhibitors (Sigma, 11836170001). Cultured cells were lysed in RIPA buffer with protease inhibitors (80 µL per 1 million cells). Then, 70 µL of cell lysates were mixed with 35 µL of 6x Laemmli sample buffer with 5% β-mercaptoethanol and boiled at 95˚C for 5 minutes. Samples were then loaded onto a 10% Tris/glycine SDS-PAGE gel (Biorad) and transferred to PVDF membranes. Membranes were blocked for 1 hour at room temperature with 3% BSA in TBST and incubated with primary antibodies against TREX1 (CST, 15107, 1:1,000), HA (CST, 3724, 1:1000) or GAPDH (CST, 2118, 1:1,000) overnight. For cell culture studies of WT TREX1 and TREX1 E266X, an N-terminal anti-HA antibody was used to probe membranes. Secondary staining was performed using horseradish peroxidase-conjugated antibodies against rabbit (CST, 7076, 1:10,000) or mouse (CST, 7074, 1:10,000) IgG. Blots were developed using Pierce ECL Western blotting substrate (Thermo, 32106) and scanned with a Bio-Rad XRS + gel imaging system.
Confocal Microscopy Studies
For confocal microscopy analysis of TREX1 localization, 293T cells with inducibly expressed HA-tagged WT TREX1 or TREX1 E266X were cultured for 2 days on poly-L-lysine-coated coverslips in complete media with doxycycline (500 ng/mL). Then cells underwent fixation with 4% paraformaldehyde for 15 minutes at room temperature. Cells were then permeabilized and blocked with blocking buffer (5% donkey serum in PBS with 0.3% Triton X-100). The cells were immunostained overnight with anti-HA antibody (CST 2367 S, 1:10000) in blocking buffer at 4˚C. Secondary staining was performed with AF647- or AF488-conjugated anti-rabbit secondary antibody for 1 hour at room temperature. Nuclei were counterstained with diamidino-2-phenylindole dihydrochloride (DAPI) and mounted in Prolong Gold anti-fade mounting medium. Images were acquired on the Leico TCS SP8 WLL Confocal and were analyzed in ImageJ.
Immunofluorescence Analysis of TREX1 in Mouse Livers
Liver tissue was excised from euthanized mice and then flash frozen in OCT-embedding media on dry ice. OCT-blocked livers were sectioned to a thickness of 8 μm by the University of Pennsylvania Molecular Pathology and Imaging Core. Frozen sections were allowed to thaw for 5 minutes at room temperature before fixation with 3% paraformaldehyde for 15 minutes at room temperature. Tissue sections were washed with 1X TBST (CST) and blocked with 1X TBST supplemented with 10% normal donkey serum. Sections were incubated with anti-HA (CST, 3724) overnight at 4 °C in a humidified chamber and visualized with fluorescent secondary antibody donkey anti-rabbit AF488 (Invitrogen). Nuclei were stained with DAPI and then coverslips applied with Prolong Gold anti-fade mounting medium. Epifluorescence images were acquired on an EVOS M5000 microscope and analyzed using FIJI software.
Screening of epegRNAs and Deep Sequencing Analysis
We began to develop a gene therapy by designing hundreds of potential prime editor gRNA (pegRNA) sequences followed by engineered pegRNA (epegRNA) sequences. Construction of epegRNA and sgRNA expression plasmids was performed as described previously [16]. Briefly, epegRNA and sgRNA oligonucleotides were designed with the assistance of prime design software [17] and synthesized by IDT. Oligonucleotides were annealed and cloned into epegRNA acceptor plasmid pU6-tevopreq1-GG-acceptor (Addgene, 174038) and sgRNA acceptor plasmid pU6-pegRNA-GG-acceptor (Addgene, 132777) digested with BsaI and assembled via Gibson assembly using T4 DNA ligase. At these initial stages, we screened 6 protospacers with 19 unique reverse transcription template (RTT) sequences. Thus, many unique epegRNAs were screened as potential prime editors of the TREX1 V235Gfs mutant. Transfected 293T cells were trypsinized and washed with 1 mL DPBS. DNA was extracted using the Qiagen DNeasy extraction kit and eluted in 100 μL elution buffer AE. The TREX1 V235Gfs cDNA was PCR amplified with the following PCR primers 5’ ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTGATGTCCTGGCCCTGCT 3' and 5’ GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCCCTTCGTCTGACGTGGCAGC 3' (Illumina adaptor sequences are underscored). For analysis of liver tissue from AAV-treated mice, livers were excised, and DNA was extracted using the Qiagen DNeasy extraction kit and eluted in 200 µL elution buffer AE. PCR amplicons were generated using the following PCR primers (Illumina adaptor sequences underscored) 5’ ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTGATGTCCTGGCCCTGCT 3’ and 5’ GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGATAGGCAGCCTGCACC 3’ NGS Amplicon sequencing was performed by Azenta, and editing efficiency was determined using the Crispresso2 web tool [18]. Editing efficiency was determined by dividing unmodified HDR reads/total aligned reads and expressed as a percentage. Indels were calculated by the sum of the modified reference and HDR reads/total aligned reads and expressed as a percentage.
Adeno-Associated Virus (AAV)
For construction of AAV genome plasmid encoding the epegRNA and sgRNA oligonucleotides were designed to PCR amplify the human U6 promoter and tevopreq motif, a second PCR was performed amplifying the desired epegRNA cassette. A third PCR reaction was performed amplifying the sgRNA cassette and mouse U6 promoter sequence. These DNA fragments were assembled with PCR amplicons encoding the AAV vector backbone derived from V3em-Cterm-PE2MAX-ΔRnaseH-dualU6 (Addgene, 198735) using Nebuilder HIFI assembly master mix (NEB) according to manufacturer’s instructions. The epegRNA sequence was: ACCATCAGGCCCATGTATGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCAGAGGCTGTGACCCCATACATGGGCCTGATCGCGGTTCTATCTAGTTACGCGTTAAACCAACTAGAA. The sgRNA sequence used in cells was: GAGGCTGTGACgccATACATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC. The sgRNA sequence used in mice was: GAGGCTGTGACCCCATACATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC. Prime editor AAV plasmids were transferred to Packgene Biotech for packaging with AAV serotype 8.
Gene Editing in mice
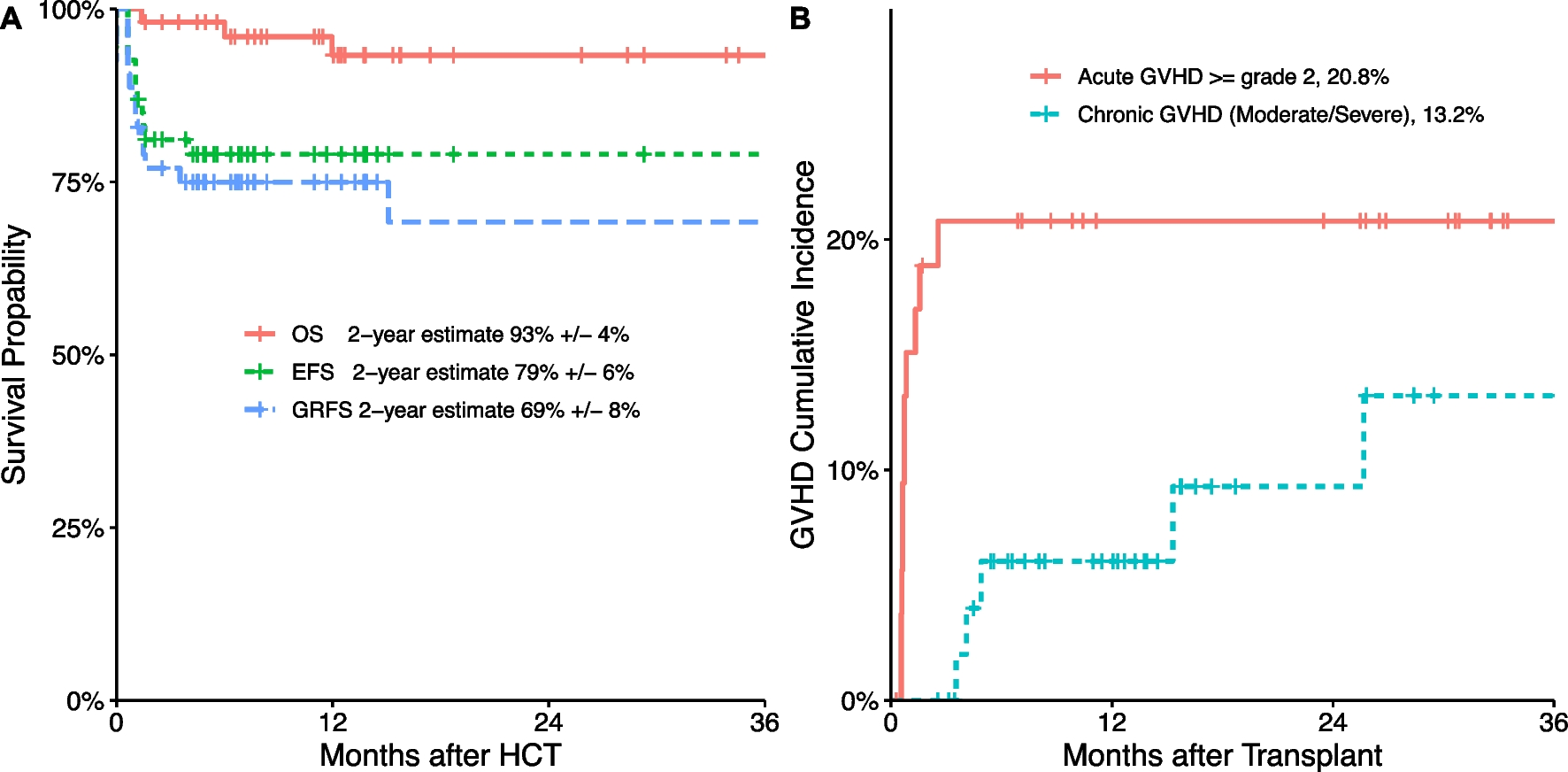
CAG-Cre LSL TREX1 V235Gfs mice were intravenously injected (via the retroorbital plexus) with 1 × 1012 genome copies (GCs) of both N- and C-terminal AAV prime editors (for a total of 2 × 1012 GCs). TREX1 expression was induced on day 18 after AAV transduction by daily intraperitoneal injection of tamoxifen (75 mg/kg) for three days. Timing was based on the fact that we have observed optimal expression of TREX1 after 4 days of induction. Liver samples were collected on day 22, andtiming of harvest after AAV administration was based on previous work using AAV gene therapies [19].
Statistics
Statistical analyses are indicated in the figure legends and were conducted in Prism (GraphPad) Version 10.4.0.




留言 (0)