
記住我




We encountered a consanguineous family with two affected children presenting with severe and recurrent respiratory infections with digestive tract symptoms (Fig. 1A, B). They were born at term after an uneventful pregnancy without any relevant family history. They had almost normal blood counts and serum immunoglobulin levels except for increased IgE (Table S1). Patient 1 was confirmed to be allergic to dust mites by a prick test at the age of 19 years.
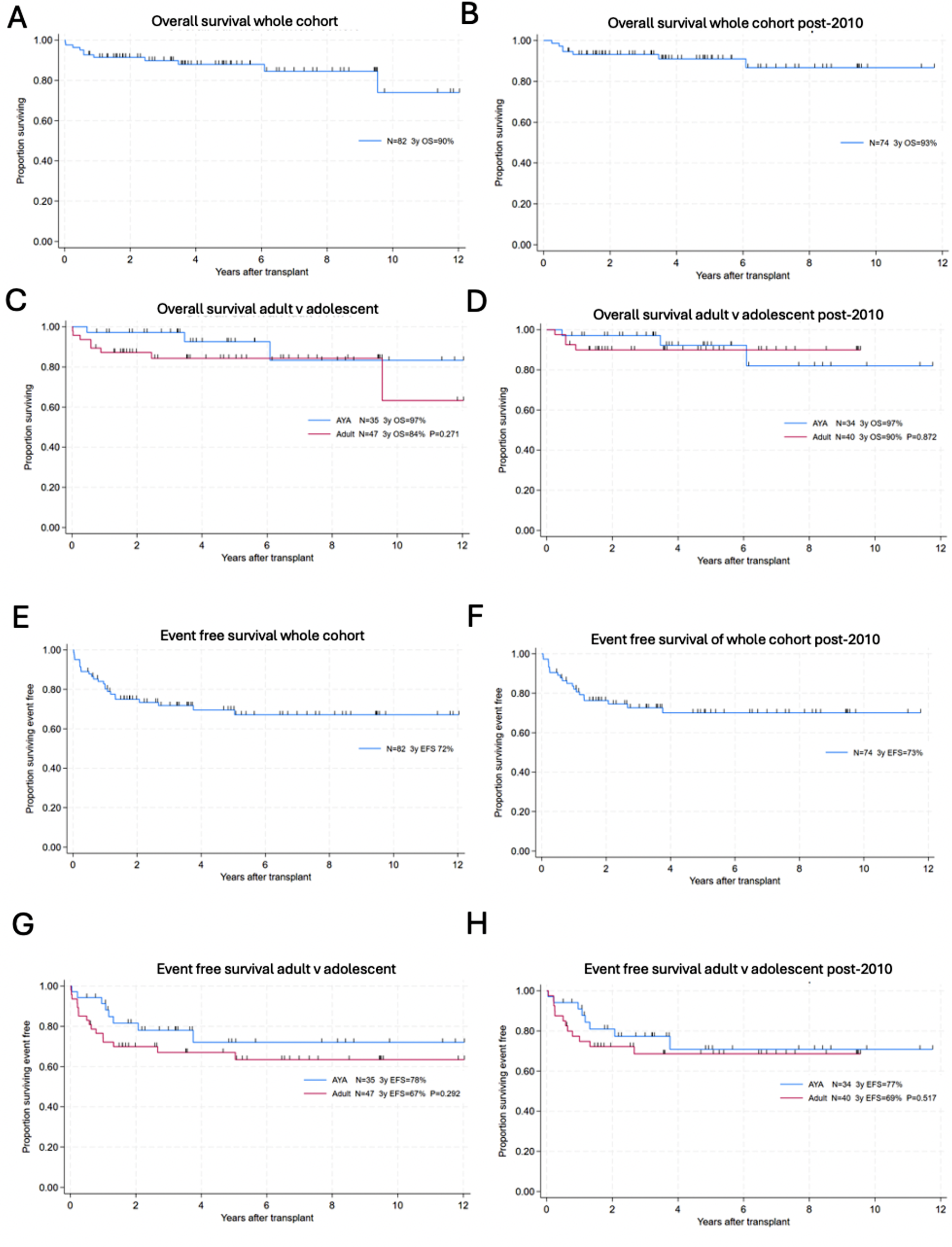
Fig. 1
Clinical features and genetic analysis of the patients. A: Pedigree tree. B: Endoscopic imaging of Patient 1 at 18 years. Mucosa is erythematous with erosions in the lower rectum and part of the anal canal (lower panel) whereas the other intestine appeared normal (upper: ileum). C: Colon histology of Patient 1 at 15 years. Crypt distortion (crypt shortening and crypt branching), cryptitis, patchy lymphoplasmacytic inflammation and decreased goblet cells were observed. D: Chromatogram of the AGR2 variant (c.250A>C, p.(Ser84Arg)). Ref: reference, Var: variant
The proband, a male patient (II-1 in Fig. 1A), presented with recurrent sinusitis and upper respiratory infections after 4 months; these persisted even after monthly treatment with intravenous immunoglobulin. He had had gastro-esophageal reflux since he was a toddler. At 2 years old, his upper digestive endoscopy revealed Grade A esophagitis according to the Los Angeles classification, petechial and erosive antral gastritis, duodenal ulcer, and lymphonodular duodenal hyperplasia. Histological examination was compatible with non-specific chronic esophagitis and gastritis. At 7 years old, he had an episode of adenovirus pneumonia complicated with pleural effusion and was diagnosed with bronchiectasis persisting until now, which was treated as bronchial asthma with inhalators. He also demonstrated bronchiectasis and presented with recurrent bacterial otitis media resulting in hearing loss at the age of 13 years. Lower digestive endoscopies at the ages of 15 and 18 years showed an erythematous colon with erosions (Fig. 1B). Colon biopsy was also performed at the age of 15 years, following 1 year of severe constipation, bloody stools, and rectal bleeding; it showed crypt distortion, cryptitis, patchy lymphoplasmacytic inflammation, and decreased goblet cells but no cryptitis, cryptic microabscess, or granulomas (Fig. 1C, and Figure S1), suggesting ulcerative colitis. At his current age of 19 years, he had normal stature (weight: 58 kg [− 1.21 SD]; height: 165 cm [− 1.62 SD]), normal secondary sexual development, and normal intellectual development (he currently attends college).
Patient 2, the younger sister of Patient 1 (II-2 in Fig. 1A), had more severe symptoms. She was born with a birth weight of 3630 g (+ 1.6 SD) and a height of 51 cm (+ 1.3 SD). She had suffered from recurrent respiratory infections with bronchial obstruction since she was 3 months old. At the age of 4 years, she presented with severe adenovirus pneumonia requiring invasive mechanical ventilation. She also suffered from recurrent and multi-drug resistance pneumonia and bronchial asthma attacks since the age of 4 years, resulting in bronchial obstruction, bronchiectasis, and atelectasis, which required domiciliary oxygen therapy. Cystic fibrosis was ruled out by sweat and chloride conductivity testing. She had been diagnosed with pulmonary hypertension at 5 years old, which had deteriorated for some time and was accompanied by tricuspid valve regurgitation, but was now well controlled with sildenafil. Although she had been treated with monthly intravenous or subcutaneous immunoglobulin since she was 7, she still presented with recurrent respiratory infections and progressive respiratory failure requiring a bi-pulmonary transplantation at the age of 14 years. After lung transplantation, the frequency of respiratory infections decreased dramatically, allowing the cessation of oxygen use. Graft function 3 years after transplantation was normal. Histological examination of the extracted lung tissues revealed bronchiolectasis with foci of chronic follicular inflammation and acute bronchiolitis, without signs of specificity (Supplementary text).
She also suffered from severe digestive symptoms. At the age of 10 years, to investigate the cause of her acute anemia and melena, upper digestive endoscopy was performed. Esophagitis (Los Angeles grade D), gastric ulcers and atrophy, and severe pyloric stenosis of 95% of the lumen with probable peptic ulcer etiology were observed. Biopsy of duodenal and gastric mucosa revealed mild villous atrophy, elongation of crypts, and lining epithelium with dedifferentiation and decreased goblet cells (Figures S2-5). She was treated with amoxicillin/clarithromycin to eradicate Helicobacter pylori, and remission was maintained with omeprazole. Her pylorus stenosis was dilatated by ballooning every 2 weeks, and she was able to eat a chopped-food diet. Her digestive symptoms were improved by changing to a gluten-free diet. At the age of 12 years, she had upper gastrointestinal bleeding. She underwent gastrostomy because of severe esophagitis and pyloric stenosis secondary to a peptic ulcer; this improved and she was able to eat orally after 6 months. She showed progressive growth impairment: her weight was 26 kg (− 1.36 SD) and her height was 128 cm (− 1.56 SD) at the age of 10 years, and her weight was 43 kg (− 1.65 SD) with a height of 146 cm (− 2.46 SD) at the age of 15 years. Currently, she is 17 years old and has had two incidences of pneumonia after a lung transplant at the age of 14 years, both of which required admission to a basic care unit but not to intensive care or requiring invasive mechanical ventilation.
Fig. 2
Functional domains and motifs of AGR2 and pathogenic variants. A: AGR2 contains a cleavable signal peptide (1–20 a.a., yellow-green box), adhesion domain (21–44 a.a, sky-blue box), dimerization domain (60–64 a.a., orange box), CXXS motif (81–84 a.a., pink box), peptide binding loop (104–111 a.a., gray box), and KTEL ER retention signal (172–175 a.a., yellow box). Previously reported variants and that found in our patients are shown in black and red, respectively. B: Evolutionary conservation of the amino acid substitution
Fig. 3
Impact of the AGR2 Ser84Arg substitution. A-B: Whole structure and magnified view of AGR241 − 175 (PDB ID: 2LNS). C: Immunoblot of V5-AGR2 transiently overexpressed in HEK293T cells under reducing and non-reducing conditions. Arrows indicate the dimer of cleaved AGR2 (dC-AGR2:36 kDa), uncleaved monomer (UC-AGR2: 21 kDa), and cleaved monomer (C-AGR2:18 kDa). Representative images are shown from three independent experiments. D: Relative expression level of V5-AGR2 under reducing conditions. E: Relative expression level of V5-AGR2 dimers under non-reducing conditions. Wild-type-cleaved monomer/β-actin = 1 (D, E). The mean and standard deviation of three independent experiments is shown. **P < 0.01 and ****P < 0.0001 analyzed by Dunnett’s multiple comparisons test (D) or ratio unpaired t-test (E)
Fig. 4
Size exclusion chromatography analysis of AGR2 variants. A: Size exclusion chromatography (SEC) profiles of AGR241 − 175 WT, Glu60Ala, and Ser84Arg eluted from a Superdex 200 10/300 column under reducing or non-reducing conditions. B: SEC profiles of AGR241 − 175 WT, Cys81Ala, and Ser84Arg under reducing or non-reducing conditions. C: Eluted fractions were analyzed using SDS-PAGE with Coomassie Brilliant Blue staining. A-C: Representative data from two independent experiments are shown
Fig. 5
Mutational effects of AGR2 on ER stress and protein stability. A: Immunoblot of V5-AGR2 transiently overexpressed in HEK293T cells under reducing conditions. Arrows indicate BiP (78 kDa), UC-AGR2 (21 kDa), C-AGR2 (18 kDa), and β-actin (40 kDa) from the top. Representative images are shown from four independent experiments. B: Immunoblot of V5-AGR2 transiently overexpressed in HEK293T cells under reducing and non-reducing conditions with treatment of DMSO only or MG123. Arrows indicate dC-AGR2 (36 kDa), UC-AGR2 (21 kDa), and C-AGR2 (18 kDa) from the top. Representative images are shown from four independent experiments
Genetic AnalysisMale and female siblings in a consanguineous family were both affected, and therefore an autosomal recessive condition was strongly suspected. Using exome sequencing, we identified a homozygous missense variant of AGR2 (NM_006408.4.:c.250A>C, p.(Ser84Arg)) in both patients as a candidate after filtration as described in Table S2. Sanger sequencing confirmed the segregation status (Fig. 1D). The exome sequencing also identified homozygous variants in EPPK1, DOC2B, and LRRC37A (Table S3). However, these three residues or bases were not evolutionally conserved. The EPPK1 and LRRC37A variants were not predicted to be pathogenic by in silico prediction tools and the DOC2B variant was not predicted to cause aberrant splicing (Figures S6-8, Table S3). Thus, at the time of the study, no evidence associated these three variants with the clinical conditions of the siblings. The AGR2 variant was absent from public control databases (gnomAD, Exome variant server, or the Human Gene Variant Database), and was predicted to be pathogenic by multiple in silico predictions (SIFT: Deleterious [0.038], PolyPhen-2: damaging [0.993], MutationTaster: disease-causing [1.0], and CADD: 26.4). CADD score/Minor allele frequency plots are shown in Figure S9 with variants in the previously reported RIFTD patients. This missense variant was located in an amino acid evolutionarily-conserved region within a CXXS motif from Danio rerio to Homo sapiens (Fig. 2A, B). In addition, our patients’ phenotype overlapped with that of RIFTD (Table 1). On the basis of the American College of Medical Genetics and Genomics/the Association for Molecular Pathology guidelines [18], this variant was classified as “Likely pathogenic” (PM2, PM3, PP1, and PP3). We think this homozygous variant is the cause of the patients’ conditions.
Table 1 Clinical summary of patients with biallelic AGR2 variantsStructural AnalysisStructural analysis was performed to investigate the effects of the missense variant. AGR2 exists in a monomer-dimer equilibrium [6], and two regions regulate its homodimerization: Glu60-Lys64 through a salt bridge [19] and Cys81 through disulfide bonding [20, 21] (Fig. 3A, B). In the mutant’s solution structure (PDB ID: 2LNS), Ser84 was located opposite to the Glu60-Lys64 dimerization domain [19] (Fig. 3A). Because the oxidation-dependent dimerization of AGR2 occurs through Cys81, the only Cys residue in AGR2 [20, 21], it is possible that the Ser84Arg substitution in the CXXS domain affects Cys81-mediated disulfide bonds.
Mutational Effects on Protein Stability and Dimerization of AGR2We transiently overexpressed V5-tagged wild-type, Ser84Arg (identified in our patient), Cys81Ala (a variant which cannot make Cys81-mediated disulfide bonds), and His117Tyr (a reported founder allele [15]) AGR2 in HEK293T cells. As previously reported for His117Tyr [14], the amount of protein produced for Cys81Ala and Ser84Arg mutants was decreased under reducing conditions (Fig. 3C, D). His117Tyr expression was not significantly reduced in our experiment. Under non-reducing conditions, dimer formation was observed only in Ser84Arg (Fig. 3C, E). Because AGR2 contains only a single cysteine (Cys81), these results indicate that the Ser84Arg variant-induced oxidation-dependent dimerization was caused by Cys81-mediated disulfide bonds.
Next, we performed size exclusion chromatography using purified recombinant proteins expressed in E. coli. Because the N-terminal signal sequence of 1–20 residues is cleavable in the ER, and the 21–40 region was unstructured and does not contribute to dimerization [19], we prepared an AGR241 − 175 construct lacking residues 1–40. As a control for the monomeric structure, we used AGR241 − 175 Glu60Ala, known to abolish the homo-dimeric structure [19], as well as AGR241 − 175 Cys81Ala as a negative control for oxidation-dependent dimerization [20, 21]. The elution profile of wild-type AGR241 − 175 exhibited an earlier peak than that of Glu60Ala (Fig. 4A, C), and had a peak consistent with the peak of Cys81Ala (Fig. 4B, C). These patterns persisted under reducing and non-reducing conditions. This indicated that AGR241 − 175 forms a Glu60-dependent dimer through a salt bridge [19] rather than binding through Cys81-mediated disulfide bonds. By contrast, AGR241 − 175 Ser84Arg displayed a broader waveform representing a larger size than the wild-type under non-reducing conditions (Fig. 4A-C, lower panel). Under reducing conditions, this broader waveform was not observed, and most of the pattern matched the peak corresponding to Glu60Ala as a monomeric structure (Fig. 4A, C, upper panel). These results suggested that in structured conditions in solution, Ser84Arg AGR2 had reduced dimerization through Glu60 under reducing conditions along with enhanced intermolecular disulfide bonding, leading to aberrant multimerization under non-reducing conditions.
Mutational Effect on ER Stress and DegradationHis117Tyr was reported to increase ER stress [14]; therefore, we investigated the potential mutational effect of Ser84Arg. However, we did not observe a significant difference in BiP expression among wild-type, Ser84Arg, or His117Tyr of transiently overexpressed AGR2 (Fig. 5A, Figure S10). However, we observed significantly decreased protein levels of Ser84Arg compared with the wild-type and His117Tyr forms. Therefore, we investigated the degradation mechanism of these AGR2 derivatives using MG132, a ubiquitin proteasome inhibitor [22]. Protein levels of uncleaved and cleaved Ser84Arg were markedly increased under MG132 treatment (Fig. 5B), suggesting that the Ser84Arg mutant is more rapidly degraded by the ubiquitin-proteasome system than other AGR2 forms.
留言 (0)