Ethics statement
The experimental procedures were approved by the Ethics Committee of Qilu Hospital of Shandong University (approval number KYLL-202203-013). Human umbilical cords were obtained from informed consenting women delivered via normal vaginal delivery in Qilu Hospital of Shandong University and processed according to the principles expressed in the Declaration of Helsinki. All animal experimental protocols were approved by the Animal Care and Use Committee of Qilu Hospital of Shandong University. All animal experiments were performed in strict compliance with the Guide for the Care and Use of Laboratory Animals.
Isolation and identification of hucMSCs
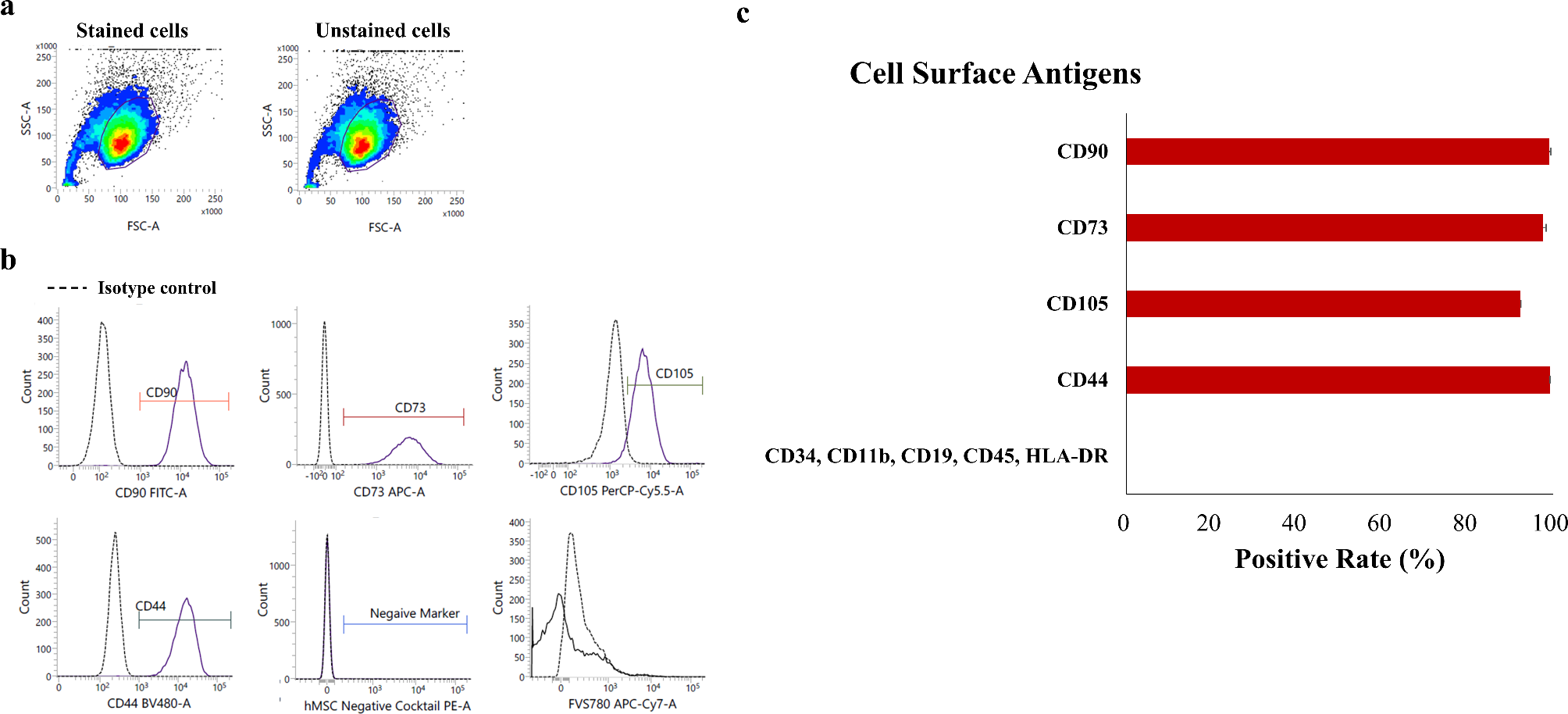
Primary hucMSCs were isolated from a fresh human umbilical cord according to the standard procedure we previously described [9, 14, 15]. After isolation, hucMSCs were maintained in alpha minimal essential medium (α-MEM, HyClone, USA) containing 10% foetal bovine serum (FBS, Gibco, USA) and 1% penicillin‒streptomycin, grown in 5% CO2 and 95% humidity at 37 °C and subcultured when they reached 80-90% confluence. The typical surface markers of hucMSCs were detected by flow cytometry using the following monoclonal antibodies: CD29, CD34, CD44, CD45, CD73, CD90, CD105, CD133, and CD146(1:1000, eBioscience, USA). Then, data were analysed using Flow Jo software. To test the multidirectional differentiation potential of hucMSCs, the cells were incubated with OriCell™ osteogenic, adipogenic, or chondrogenic differentiation media (Cyagen, China). The chondrogenic, adipogenic and osteogenic differentiation potential of hucMSCs was estimated with Alizarin Red staining, Oli Red O staining and Alcian Blue staining, respectively.
Isolation and identification of ROMECs
Rat ovarian microvascular endothelial cells (ROMECs) were isolated from fresh rat ovarian tissues using the Percoll density gradient centrifugation method based on the protocol described by Toshihiro Sakurai et al. [16]. The isolated cells were cultured in endothelial cell medium (ECM) supplemented with 5% FBS, 1% endothelial cell growth supplement, and 1% penicillin‒streptomycin in 5% CO2 and 95% humidity at 37 °C. ROMECs were detected by their cobblestone-like morphologic characteristics and CD31, CD34, and von Willebrand Factor (VWF) cell immunofluorescence staining as described [17].
Exposure of hucMSCs to hypoxia
To induce a hypoxic environment, hucMSCs were cultured in a humidified hypoxia modular incubator chamber at 37 °C with a hypoxic gas mixture composed of 5% O2, 5% CO2, and balanced N2 for 24 h. The control hucMSCs were grown in a humidified incubator at 37 °C with 5% CO2 under normoxic conditions of 21% O2.
Purification of norm-exos and hypo-exos
After 72 h incubation of hucMSCs, the culture medium supernatant of 10^8 hucMSCs cultured in normoxic and hypoxic environments were collected for norm-Exo and hypo-Exo isolation, respectively. The culture medium supernatant was centrifuged at 1500 × g for 5 min and then 10,000 × g for 20 min at 4 °C to remove debris and apoptotic bodies. This step was followed by a repeated ultracentrifugation step at 120,000 × g for 70 min at 4 °C to purify exosomes. The final exosomal pellet was resuspended in phosphate-buffer saline (PBS) and then stored at -80 °C for subsequent experiments.
Characterization of norm-exos and hypo-exos
For exosome characterization, transmission electron microscopy (TEM; JEOL-1200EX, Japan) analysis was performed to visualize the appearance of freshly purified exosomes. Nanoparticle tracking analysis (NTA) was performed to detect the size distribution and mean diameter of exosomes by laser scattering microscopy (LSM, Zeta View® x30, Germany). Specific exosomal markersCalnexin, TSG101, CD9, CD63, and CD81(1:1000, Proteintech, China)were detected by western blotting. In addition, a bicinchoninic acid (BCA) protein assay was applied to evaluate the protein concentrations of exosomes.
Labelling and tracking of norm-exos and hypo-exos
To demonstrate the uptake of norm-Exos and hypo-Exos in ROMECs, exosomes were labelled with green fluorescent dye (PKH67). ROMECs were cocultured with norm-Exos and hypo-Exos at a concentration of 25 µg/mL for 12 h. Subsequently, the cells were fixed, counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and observed using a fluorescence microscope.
EdU assay
EdU assays and colony formation assays were applied to assess cell proliferation under different conditions according to the standard protocol. For the EdU staining assay, the EdU Cell Proliferation Kit (RiboBio, China) was used to identify proliferative ROMECs following the manufacturer’s procedures. ROMECs were cocultured with norm-Exos and hypo-Exos at a concentration of 25 µg/mL for 24 h. Nuclei were stained with DAPI, and the proliferation rate of ROMECs was assessed by calculating the number of EdU-positive cells under a fluorescence microscope.
Colony formation assay
For the colony formation assay, eight hundred ROMECs were seeded into each well of 6-well plates and treated with25 µg/mL norm-Exos or hypo-Exos at a concentration of for 24 h. for 7 days to allow colony growth. Cells were fixed with formaldehyde and stained with crystal violet. The number of colonies was counted manually, and the colonies were photographed.
Wound healing assay
Wound healing assays and Transwell assays were applied to assess cell migration under different conditions in accordance with methods reported in our previous studies [14]. For the wound healing assay, ROMECs were cultured in a 6-well plate. When cell confluence reached 90%, a scratch was made in the confluent monolayer using a pipette tip. The plates were washed to remove the debris and then incubated with 25 µg/mL norm-Exos and hypo-Exos for 24 h. The wound borders were photographed at 0 h and 24 h post-scratch using an inverted microscope.
Transwell migration assay
For the Transwell migration assay, ROMECs were counted and cultured in the upper chamber of the Transwell chamber (8.0-µm pore size, Corning, USA). After cocultured with norm-Exos and hypo-Exos at a concentration of 25 µg/mL in an incubator at 37 °C and 5% CO2 for 24 h, the chamber was fixed with paraformaldehyde and stained with crystal violet. The cells that remained on the upper surface did not migrate and were removed, and those on the lower surface were visualized and imaged under an inverted microscope.
Tube formation assay
The formation of vessel-like structures was examined in a 24-well plate using an In Vitro Angiogenesis Assay Kit (Abcam, USA) following the manufacturer’s instructions. ROMECs under different culture conditions were plated on extracellular matrix gels and incubated with 25 µg/mL norm-Exos or hypo-Exos at 37 °C for 12 h to allow tube formation. At the end of the cultivation, the tube network was stained with calcein acetoxymethyl, and the capillary-like structures were imaged using fluorescence microscopy. Additionally, the total tube length and tube number in three random fields were quantified as the tube formation ability.
miRNA high-throughput sequencing
Total RNA of exosomes was extracted from norm-Exos and hypo-Exos using TRIzol Reagent (Thermo Fisher, USA). RNA quantification was verified by a NanoDrop ND-1000, and RNA integrity and gDNA contamination were evaluated by denaturing agarose gel electrophoresis. The TruSeq Small RNA Sample Preparation Kit (Illumina, USA) was utilized to construct small RNA libraries. The samples were processed for deep sequencing using a MiSeq desktop sequencer system (Illumina, USA) according to the manufacturer’s instructions. An adjusted P value < 0.05 and |log2(fold change)| > 1 indicated significantly differentially expressed genes. Prediction of downstream genes regulated by miRNAs was performed using microRNA Data Integration Portal (mirDIP), miRwalk, starBase, TargetScan, and miRDB.
Dual luciferase reporter assay
The binding sites between PTEN and miR-205-5p were predicted by a biological prediction website and further validated by the dual luciferase reporter assay. The mutant-type (MUT) 3′-UTR (MUT-PTEN) and wild-type (WT) 3′-UTR (WT-PTEN) were synthesized into the pmirGLO dual luciferase reporter vector (Promega, USA). Then, ROMECs were transfected with miR-205-5p NC or miR-205-5p mimic and later co-transfected with the two reporter plasmids according to our previously described protocol [18]. After 48 h, luciferase activities were measured using the Dual Luciferase® Reporter Assay System (Promega, USA) according to the manufacturer’s instructions.
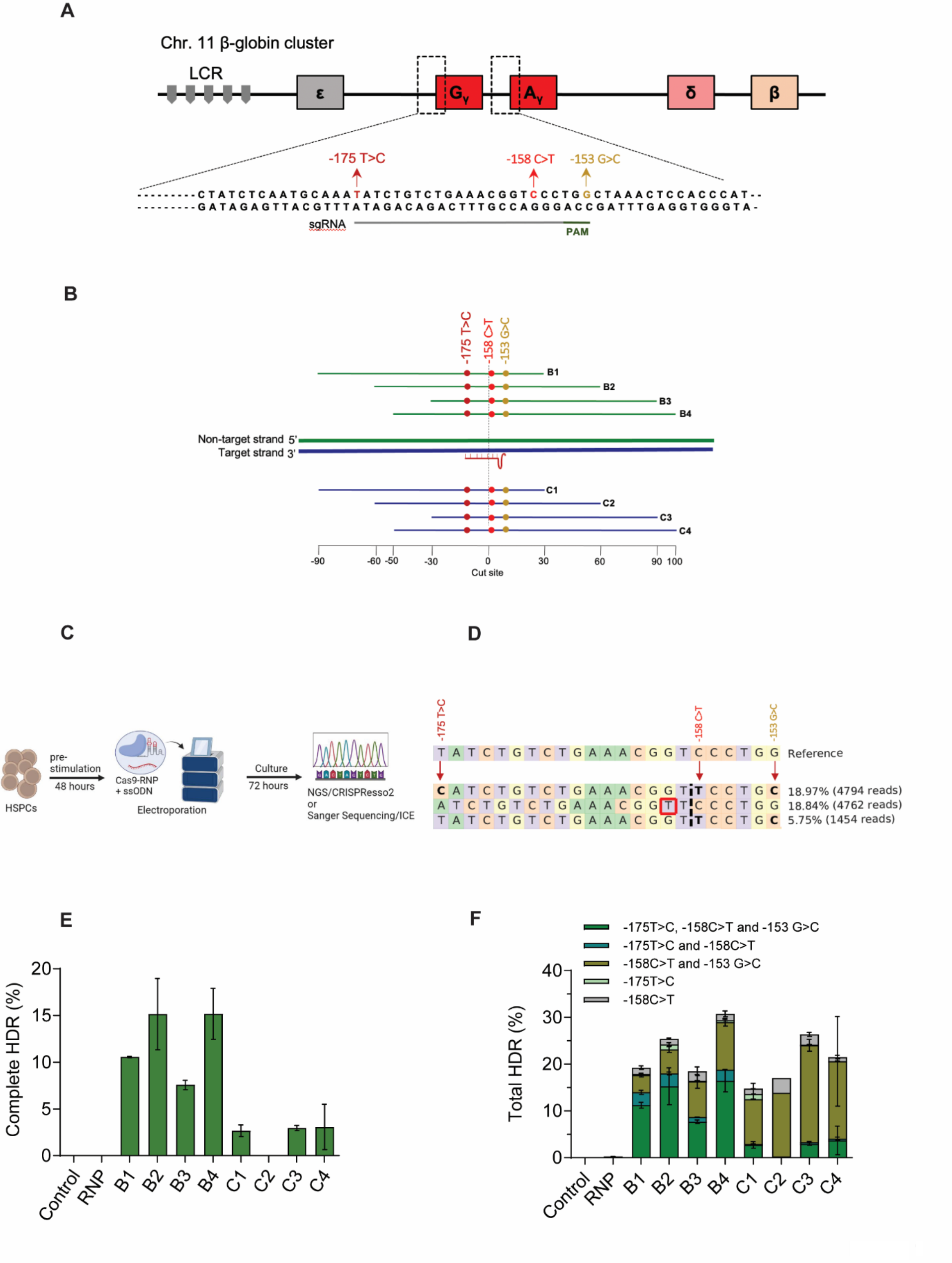
Establishment of the POF rat model
The work has been reported in line with the ARRIVE guidelines 2.0. Female Wistar rats weighing 170–190 g were obtained from Pengyue Laboratory Animal Co., Ltd. (Jinan, China) and raised in a pathogen-free environment. To establish the rat POF model, Wistar female rats were injected intraperitoneally with cisplatin for 14 days based on a protocol we described previously [9]. Cisplatin-induced POF rats were intravenously injected with a single dose of exosomes (400 µg dissolved in 200 µL PBS) obtained from different cell conditions under conscious state. and then equally and randomly divided into groups for different administration after modelling. The POF rats were weighed, and serum levels of follicle-stimulating hormone (FSH), oestradiol (E2), and Anti-Müllerian hormone (AMH) were detected by enzyme-linked immunosorbent assay (ELISA) at 0, 1, 2, and 4 weeks after exosome treatment. At the end of the study, these rats were euthanized by phenobarbitone and euthanized by carbon dioxide, and the ovaries were removed for subsequent experiments. The ovarian structure and follicle development in POF rats were determined by haematoxylin and eosin (HE) staining, proliferating cell nuclear antigen (PCNA) immunohistochemistry staining and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining. The angiogenesis status of ovarian tissues in POF rats was determined using CD31 immunofluorescence staining.
Ovarian follicle counting and morphological analysis
The ovarian tissues were fixed with formalin, dehydrated in ethanol, cleared in xylene, embedded in paraffin, and sliced into 5-µm-thick sections. The slides were stained with HE using a Haematoxylin-Eosin Stain Kit (Solarbio, China) to detect ovarian morphology and follicle counts in accordance with the manufacturer’s instructions. The different categories of follicles, including atretic, primordial, primary, and secondary follicles, were counted by two investigators as described in the literature.
Immunohistochemical staining
For immunohistochemical staining, paraffin-embedded ovarian tissue sections were dewaxed in xylene, rehydrated through graded ethanol, heated in ethylenediaminetetraacetic acid (EDTA) solution, and blocked with bovine serum albumin (BSA). Next, slides were incubated with diluted PCNA primary antibody (1:500, Abcam, USA) followed by incubation with peroxidase-labelled secondary antibodies. PCNA-positive signals were finally observed with 3,3′-diaminobenzidine solution by counterstaining with haematoxylin reagent.
Immunofluorescence staining
For immunofluorescence staining, tissue samples from the in vivo experiment were blocked with 5% goat serum, incubated with diluted CD31 primary antibody (1:200, Abcam, USA), and stained with rhodamine–conjugated secondary antibodies. Next, tissues were counterstained with DAPI, and the stained sections were visualized by fluorescence microscopy to evaluate microvessel density in ovarian tissue. For cell immunofluorescence to identify primary ROMECs isolated from rat ovarian tissues, the antibodies used in this study were as follows: anti-VWF-FITC (1:100, Abcam, USA), anti-CD31 (1:200, Affinity, China) and anti-CD34 (1:200, Abcam, USA).
Western blotting
For western blot analysis, total protein from cell samples was isolated using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, China), and the protein concentration was measured by using a Bicinchoninic Acid (BCA) Protein Assay Kit (Beyotime, China). Proteins were separated by sodium dodecyl sulfate‒polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, USA). Then, the membranes were blocked with nonfat milk and incubated with primary antibodies. Next, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000, Proteintech, China). The resulting protein bands were visualized by the enhanced chemiluminescence detection system (Millipore, USA) The main antibodies used in this study are as follows: Calnexin (1:2000, Affinity, China), TSG101 (1:2000, Proteintech, China), CD9 (1:1000, Proteintech, China), CD63 (1:1000, Proteintech, China), CD81 (1:1000, Proteintech, China), GAPDH (1:5000, Proteintech, China), PTEN (1:2000, Affinity, China), PI3K (1:2000, Cell Signaling Technology, USA), p-PI3K (1:2000, Cell Signaling Technology, USA), AKT (1:2000, Cell Signaling Technology, USA), p-AKT (1:2000, Cell Signaling Technology, USA), mTOR (1:2000, Cell Signaling Technology, USA), and p-mTOR (1:2000, Cell Signaling Technology, USA).
Quantitative real‑time polymerase chain reaction (qRT‑PCR)
For qRT‑PCR analysis, total RNA of cell or tissue samples was extracted using TRIzol Reagent (Thermo Fisher, USA). The cDNAs were synthesized by using a miScript II RT Kit (Qiagen, Germany), and then qRT‒PCR was performed using a miScript SYBR Green PCR Kit (Qiagen, Germany) in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA) according to the manufacturer’s instructions. miRNA and mRNA expression levels were calculated using the 2 − ΔΔCt method with U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the endogenous controls.
Statistical analysis
Data are reported as the means ± standard deviations (SD), and statistical analyses were performed using SPSS 22.0 software (SPSS, USA) and GraphPad Prism 6.0 software (GraphPad, USA). One-way analysis of variance (ANOVA) followed by Student-Newman‒Keuls (SNK) test was applied to analyse variance among all groups. At least three biological replicates were performed for every experiment in this study, and a P value < 0.05 was considered statistically significant in the present study.






留言 (0)