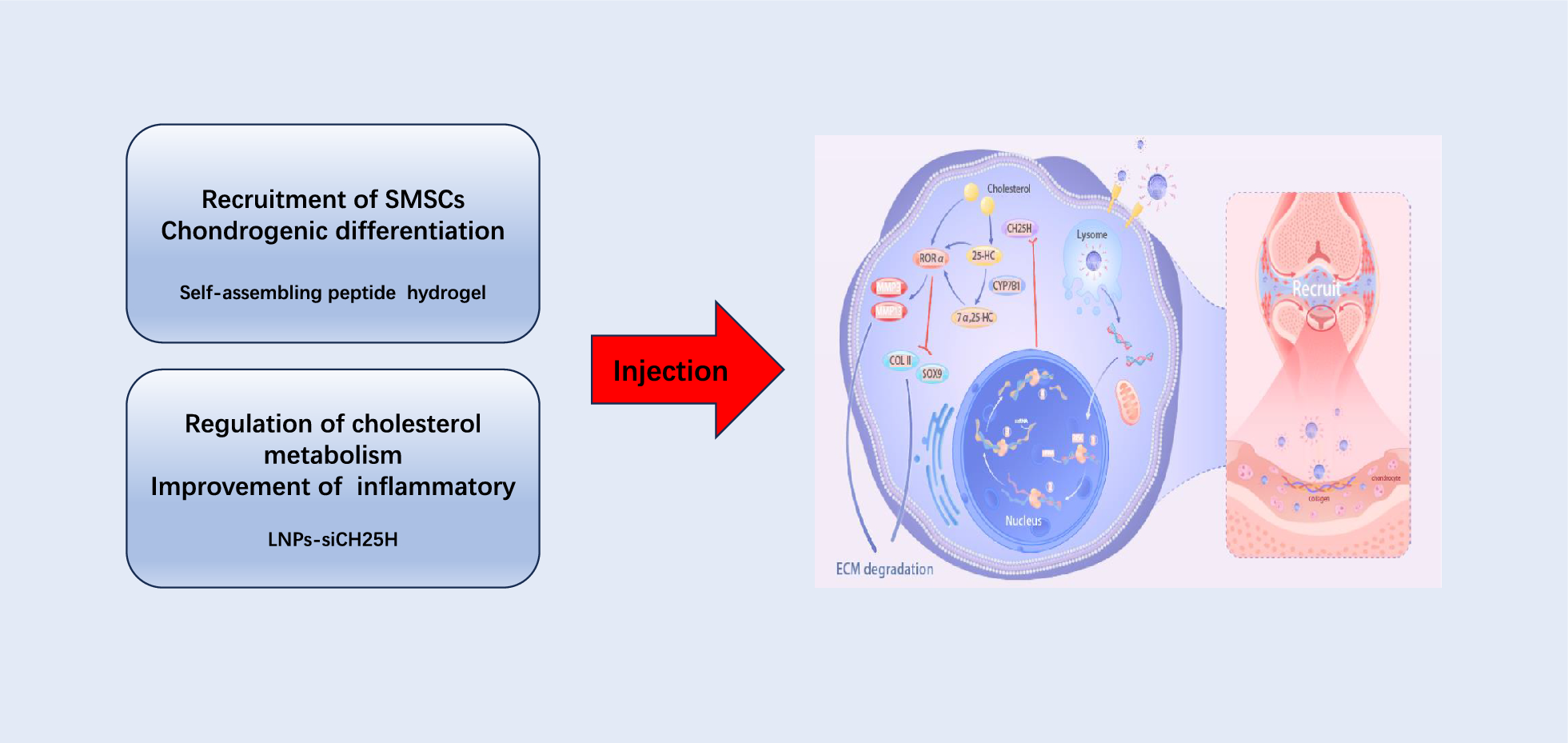
Material characteristicsPreparation of the peptide hydrogel
The peptide sequences of the self-assembling peptides Fmoc-K(Fmoc)SKPPGTSS and Fmoc-LIANAK (purity > 95%) were synthesized and purified by China Peptides (Shanghai, China) via the solid phase peptide synthesis (SPPS) as previously described [71, 72]. In short, 1 mmol of resin was weighed and placed in a reactor, dichloromethane (DCM) was added to swell for half an hour, then DCM was removed, the first amino acid (2 mmol) in the sequence, 2 mmol of N, N-diisopropylethylamine (DIEA), an appropriate amount of dimethylformamide (DMF) and DCM was added, and then nitrogen bubbling was reacted for 60 min. Then 5 mmol of methanol was added, reacted for half an hour, the reaction liquid was removed, and washed with DMF and methanol. Then the second amino acid in the sequence (2 mmol), 2 mmol of 1-hydroxybenzotrichloridazole tetramethyl hexafluorophosphate (HBTU) and DIEA were added to the reactor, nitrogen bubbling was reacted for half an hour, the liquid was washed away, ninhydrin was detected, and then pyridine and acetic anhydride were used for end-capping, and finally washed, an appropriate amount of decapping liquid was added to remove the Fmoc protecting group, washed, and ninhydrin was detected. Different amino acids in the sequence were added in sequence, and various modifications were performed in the same way as before. The resin was then dried with nitrogen and taken out from the reaction column, poured into a flask, and then a cutting solution (95% trifluoroacetic acid, 2% ethanedithiol, 2% triisopropylsilane, and 1% water) was added to the flask, shaken well, and the resin was filtered out. Finally, a large amount of ether was added to the filtrate to precipitate the crude product, which was then centrifuged and washed to obtain the crude product. The crude product was purified by HPLC to a purity of more than 95%, then concentrated by a freeze dryer and lyophilized into a white powder. The purity and successful synthesis of the target peptides were verified by HPLC and ESI–MS analysis. Based on the peptide hydrogel self-assembly synthesis method in previous studies, we used solvent switching technology to make the peptide gel [71, 73, 74]. Tests found that when the peptide solution concentration reached 3.5% (w/v), the hydrogel was able to self-assemble (Fig. S2). Therefore, the final concentration of all hydrogels in this study was set to 3.5% (w/v). Briefly, Fmoc-K(Fmoc)SKPPGTSS (5 mg) and Fmoc-LIANAK (5 mg) were co-dissolved in DMSO at a concentration of 160 mg/mL. Afterward, these peptide stocks were diluted with ultrapure water to a final concentration of 35 mg/mL to form a gel. For in vitro cell studies, ultrapure water sterilized by UV light was used to prepare the hydrogels. The peptide stock solution (350 µL per well in a 24-well plate) was loaded into the wells and sterilized with UV light for 1 h. Subsequently, sterile ultrapure water was added to the hydrogel to gel. The plate was then incubated (37 °C, 5% CO₂) to allow the hydrogel to equilibrate before seeding the cells.
Preparation and characteristics of the C5-24-LNPs-siRNA
The LNPs-siRNA was synthesized by Genenc Biomedical Technology (Hangzhou) Co. Ltd. The LNPs-siRNA were fabricated via a self-assembly approach employing the ethanol dilution technique [75,76,77]. Dlin-MC3-DMA, DSPC, cholesterol, DMG-PEG2000, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[(polyethylene glycol)-2000]-maleimide (DSPE-PEG2000-MAL) were purchased from AVT (Shanghai) Pharmaceutical Tech Co., Ltd. The siRNA was synthesized by Tsingke Biotechnology Co., Ltd. Initially, Dlin-MC3-DMA, DSPC, cholesterol, DMG-PEG2000, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[(polyethylene glycol)-2000]-Maleimide (DSPE-PEG2000-MAL) were mixed and dissolved in ethanol at a ratio of 50:10:38:1.5:0.5. Simultaneously, the siRNA was diluted in a citrate buffer solution, which had been prepared to a concentration of 10 mM and adjusted to a pH of 4. Following this preparation, the ethanol phase containing the lipids was rapidly mixed with the aqueous siRNA solution. The mixing was conducted at a volume ratio of 1:3 (ethanol to aqueous) and maintained at a total flow rate of 4 mL/min. This rapid mixing allowed the lipids to self-assemble into nanoparticles encapsulating the siRNA. The resulting mixture was then incubated at room temperature for a duration of 10 min to ensure complete nanoparticle formation. To purify the RNA-loaded lipid nanoparticles, the mixture was transferred to a Millipore centrifuge tube and subsequently centrifuged at 2500 rpm. After centrifugation, the nanoparticles underwent three cycles of ultrafiltration to remove any unencapsulated siRNA and residual solvents, resulting in the final purified LNPs-siRNA preparation. Then, the C5-24 peptide (CDLQYWYPIWDTH) was synthesized by ChinaPeptides (Shanghai, China) through solid phase synthesis. The crude product was refined to over 90% purity using high-performance liquid chromatography. The purified solution was then concentrated in a freeze dryer and lyophilized into a white powder. The C5-24 peptide underwent HPLC and ESI–MS analyses to confirm the target peptide's purity and validate the synthesis process. Meanwhile, the thiol group of cysteine in C5-24 peptide was conjugated to DSPE-PEG2000-Mal of LNPs surface to form C5-24-PEG2000-DSPE [68]. Finally, C5-24-PEG2000-DSPE micelles were incorporated into the surface of LNPs. Dynamic light scattering (DLS) was utilized to assess the particle size of the nanoparticles following preparation and encapsulation. The Zeta potential was measured using a specialized analyzer based on the electrostatic double-layer model at the solution interface, as described by Stern's theory. The hydrodynamic diameter and zeta potential were measured by laser light scattering using a Litesizer 500 (AntonPaar, Graz, Austria). Finally, the microstructure of C5-24-LNPs-siRNA was observed by cryo-TEM. The encapsulation efficiency of siRNA within the LNPs was determined using a fluorescence-based assay.
CD analysis of the peptide
Secondary structure was analyzed by CD using a CD spectrometer J-810 spectropolarimeter (JASCO Corporation, Tokyo, Japan). To reduce scattering, dilute the sample to about 0.01% (w/v) with pure water. Approximately 400 µL of the diluted sample was transferred to a cuvette with a path length of 10 mm. CD spectra were acquired from 180 to 320 nm with a step size of 0.5 nm and the baseline was subtracted. The obtained data were averaged and smoothed using software for further analysis.
FTIR spectroscopy
FTIR spectra of the peptide hydrogel were acquired by depositing 30 µL samples onto KBr infrared cards, drying them under vacuum conditions, and analyzing them using an FTIR spectrometer (Nicolet iS50; Thermo Fisher Scientific, Waltham, MA). This method allowed detailed examination of the chemical structure and composition of the hydrogel.
Rheology
The rheological properties of the peptide hydrogel were investigated using an RS75 rheometer (Haake, Germany) equipped with an 8-mm diameter parallel plate geometry. The gel was subjected to a strain of 0.5%, and stress–frequency curves were generated across frequencies ranging from 0.1 to 10 Hz. Additionally, the viscosity–shear rate relationship was assessed logarithmically over shear rates from 0.01 to 100 1/s. Storage modulus (G’) and loss modulus (G”) were determined and plotted for comprehensive analysis. Each experiment was conducted in triplicate to ensure accuracy and reliability of the results.
TEM
The 10 mg/mL peptide solution was deposited onto a copper grid for TEM analysis using a JEM-2100F microscope (JEOL, Tokyo, Japan). Images were captured to observe the nanostructure of the peptides in detail.
Cryo-TEM
The size, morphology, and distribution of the LNPs were confirmed by cryo-TEM. For cryo-TEM examination, the temperature of the FEI Vitrobot chamber was first set to 4 °C, the relative humidity was set to 100%, a special filter paper (Whatman grade 1) was inserted, and the system was allowed to stabilize for 10 min. Then, the clamping parameters were configured with a clamping time of 3 s, a clamping force of 10 N, and a waiting time of 30 s. Liquid nitrogen was poured into the cryogen container of the Vitrobot and allowed to stabilize for 5 min to achieve a consistent low temperature. Ethane gas was then injected into the container to liquefy it to a level of approximately two-thirds of the height of the copper cup. A copper grid (Quantifoil R2/2) was hydrophilized using a glow discharge device (PELCO easiGlow) at 15 mA for 60 s. The hydrophilized copper grid was placed in the Vitrobot chamber. Here, 3 µL of the sample solution was dropped onto the grid and the sample was allowed to sit for 30 s to allow the particles to fully adsorb to the grid surface. The grids were then blotted for 2–4 s with filter paper and immediately immersed in liquid ethane after removing excess sample solution with filter paper. For subsequent examination, the samples were transferred to a TEM (FEI Talos F200C, Thermo Fisher Scientific, Naarden, The Netherlands) and kept under cryogenic conditions. Conventional TEM imaging was performed using an accelerating voltage of 300 kV. Micrographs were taken under low-dose conditions using a 4 k direct electron detection camera (Gatan K3, Pleasanton, CA, USA). Images were subsequently analyzed using ImageJ software (version 1.54).
SEM
Following gelation, the peptide hydrogel underwent a 24-h freeze-drying process in a lyophilizer. Subsequently, the lyophilized samples were meticulously examined and imaged under a SEM model S4800 by HITACHI, located in Tokyo, Japan.
Gel retardation assay
Gel retardation assay was used to investigate the encapsulation effect of LNPs with siRNA. NPs were formulated with lipid and siRNA at the following ratios: 4:1, 8:1, 16:1, 24:1, and 32:1. 10 µL aliquots containing 0.2 µg of siRNA were mixed with RNA loading buffer and introduced into parallel wells of a 3% (w/v) agarose gel. The gel also included 1 µL/mL GelRed dye (Beyotime, Shanghai, China) in Tris–acetate-EDTA buffer. Electrophoresis was performed at 120 V for 60 min, and the gel retardation pattern was captured using an Amersham Imager 600 RGB (Amersham plc, UK).
In vitro experimentsPrimary articular chondrocyte and SMSCs culture
Primary articular chondrocytes were extracted from the femoral condyles and tibial plateaus of 3-day-old male Sprague–Dawley rats. Articular cartilage was removed, minced, and initially digested with 0.25% trypsin–EDTA at 37 °C, shaking at 200 rpm for 1 h, then further digested overnight with 0.2% type II collagenase at 37 °C. The mixture was filtered and centrifuged to obtain the cell pellet. The cells were rinsed twice with phosphate-buffered saline (PBS) and then cultured in DMEM enriched with 10% fetal bovine serum (KeyGen BioTECH, China) and 1% penicillin–streptomycin (KeyGen BioTECH) at 37 °C in a 5% CO2 humidified environment. Upon reaching 70–80% confluency, the cells were exposed to murine IL-1β (Beyotime, China) to initiate inflammation. The primary articular chondrocytes were isolated from cartilage tissue using an enzymatic digestion process, as previously described. The primary SMSCs were purchased from Procell Life Science & Technology Co. Ltd. (Wuhan, China) and cultured in our laboratory following the supplier's recommended protocols. In all cell experiments, chondrocytes were used in P1, and SMSCs were used in P3. Hydrogels were prepared as described previously. Hydrogels with a diameter of approximately 6.4 mm and a height of 2 mm were formed in standard 24-well culture plates and equilibrated with culture medium in an incubator (37 °C, 5% CO₂) for at least 1 h prior to cell seeding. Articular chondrocytes and SMSCs were harvested from their respective culture flasks using standard trypsinization followed by centrifugation. The cells were then resuspended in their respective culture media at a concentration of 1 × 106 cells/mL. The prepared hydrogels in the standard 24-well culture plates were gently aspirated to remove any excess medium. For each well, 100 µL of the cell suspension (containing 105 cells) was meticulously pipetted onto the hydrogel surface. The plates were then gently agitated to facilitate uniform cell distribution across the hydrogel. The cells were allowed to adhere to the hydrogel for 2 h in the incubator (37 °C, 5% CO₂). After the adhesion period, 500 µL of the respective culture medium was gently added to each well to cover the hydrogel and cells. The plates were then returned to the incubator for further culture.
Live/dead cell assay
To investigate the biocompatibility of the hydrogels, we performed the live/dead cell assay. After 2 days of culture, cells were stained with a Calcein/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime, China) following the manufacturer's instructions. Cells were washed three times with PBS, treated with Calcein AM/PI solution, incubated at 37 °C for 30 min, rinsed again with PBS, and then examined under a fluorescence microscope (Carl Zeiss, Jena, Germany).
Cell proliferation assay
To investigate the biocompatibility of the hydrogels, we performed the cell proliferation assay. On days 1, 3, and 5 after seeding rat chondrocytes and SMSCs on the hydrogel, they were incubated with 200 μL of Cell Counting Kit-8 (Beyotime, China) solution for 2 h at 37 °C in darkness. Then, 100-μL samples of the supernatants were moved to a 96-well plate for absorbance measurement at 450 nm using a Multimode Plate Reader. To evaluate the dose-dependent toxicity of LNPs, cell proliferation assays were conducted. Primary chondrocytes and SMSCs were cultured in suitable media under standard conditions (37 °C, 5% CO2) and seeded in 96-well plates, ensuring uniform cell density across all wells. Serial dilutions of LNPs-siRNA were prepared, covering a dose range from 50 nM to 3 µM. The cells were then treated with the respective concentrations of LNPs-siRNA and incubated for 2 days. Following the incubation, 200 μL of Cell Counting Kit-8 (Beyotime, China) solution was added and the cells were incubated for an additional 2 h at 37 °C in the dark. Subsequently, 100 μL of the supernatant was transferred to new 96-well plates, and the absorbance at 450 nm was measured using a multi-mode plate reader.
Cytoskeletal staining
To study cell adhesion on the hydrogel, rat chondrocytes and SMSCs were cultured for 3 days and then subjected to cytoskeletal staining. After removing the culture medium, cells were gently washed three times with PBS and fixed with 4% paraformaldehyde (Beyotime, China) for 30 min. Following another PBS wash, cells were permeabilized with 0.1% Triton X-100 (Beyotime, China) for 5 min, blocked with 1% BSA for 30 min at 37 °C, and treated with rhodamine-phalloidin for 30 min at room temperature. Finally, the cells were washed, stained with DAPI, and observed under a confocal laser scanning microscope (M7000).
SMSCs migration assay
To investigate the SMSCs recruitment capacity of hydrogels, we performed SMSCs migration experiments. Migration tests for SMSCs were performed using transwell inserts with polycarbonate films (BIOFIL). Hydrogel was placed in the lower chamber, and SMSCs were seeded on the inserts. Culture medium was added above and below the inserts. After 24 h at 37 °C, non-migrated cells on the insert's top were removed. Migrated cells on the bottom were fixed with methanol and stained with 0.1% crystal violet. The cells were then photographed and counted under a light microscope.
SMSCs differentiation assay
To investigate the cartilage differentiation-promoting function of hydrogels, we conducted SMSCs differentiation assay. Chondrocyte differentiation was induced using TGF-β in mesenchymal stem cell differentiation mediums. Briefly, 1 × 105 SMSCs were placed on the surface of different hydrogels with 500 μL of cartilage-inducing solution and incubated at 5% CO2 for 14 days, refreshing the medium every two days. On the 14th day, the cells underwent qRT-PCR for cartilage gene expression, western blot for protein levels, Alcian Blue, and immunofluorescence staining.
Cell uptake
For the experiment to observe whether LNPs can successfully deliver siRNA to chondrocytes to avoid lysosomal degradation. First, cells were seeded in 24-well plates (3 × 104 cells/well) and induced with IL-1β for 6 h. After successful induction, the medium was aspirated and washed twice with PBS, and then an equal amount of new medium was added. The next day, cells were incubated with free Cy5-labeled siRNA or LNPs encapsulated with Cy5-labeled siRNA. The dose was 3 μg siRNA per well (30 ng/104 cells), treated at 37 °C for 1 h, then washed with PBS, observed under a confocal laser scanning microscope (CLSM), stained with DAPI (blue), and then subjected to CLSM analysis (CLSM 780, Carl Zeiss Inc.). For flow cytometry experiments, cells were seeded in 24-well plates (3 × 104 cells/well) and induced with IL-1β for 6 h. After successful induction, the medium was aspirated and washed twice with PBS, and then an equal amount of new medium was added. On the next day, cells were treated with Lipo3000 (encapsulating Cy5-labeled siRNA), LNPs without targeting peptide (encapsulating Cy5-labeled siRNA), and LNPs with targeting peptide (encapsulating Cy5-labeled siRNA) at a dose of 3 μg siRNA per well (30 ng/104 cells) at 37 °C for 1 h and then washed with PBS. To remove nonspecifically bound nanoparticles, cells were washed twice with PBS at room temperature and collected in flow cytometry buffer (PBS containing 10% v/v FBS) containing DAPI viability dye. Readings were performed in live cells using a CytoFLEX S flow cytometer (Beckman, USA). Data were analyzed using FlowJo software (version 10.10).
Immunofluorescence
Chondrocytes and SMSCs attached to the gels were fixed in 4% paraformaldehyde for 30 min, 3 μm thick sections were cut, permeabilized with 0.1% Triton X-100 for 5 min, and blocked with 1% BSA for 1 h at 37 °C. They were then incubated with primary antibodies (all primary antibodies were diluted at 1:200) overnight at 4 °C, followed by 1-h incubation at 25 °C with goat anti-rabbit IgG H&L (Alexa Fluor 594; 1:200; AB150084; Abcam) and goat anti-mouse IgG H&L (Alexa Fluor 488; 1:200; AB150117; Abcam). After DAPI staining, cells were visualized using a confocal laser scanning microscope (Celldiscoverer 7).
qRT-PCR
Total RNA was extracted and purified from chondrocytes and SMSCs attached to the gels using the SteadyPure Universal RNA Extraction Kit II (AG21022; Accurate Biology, China). The mRNA was reverse-transcribed to cDNA using the Evo M-MLV RT Kit with gDNA Clean for cPCR (AG11705; Accurate Biology). qRT-PCR was conducted using the SYBR Green Premix Pro Taq HS qPCR Kit IV (AG11746; Accurate Biology) on a LightCycler 480 real-time PCR system (Roche, USA). Target gene expression was normalized to β-actin gene expression. The 2-ΔΔCt method calculated relative expression levels, with all experiments conducted in triplicate. The primers used are listed in Table S4.
WB
Cells were washed twice with chilled PBS and lysed with cold lysis solution for protein extraction. Proteins were separated using 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA). The membranes were blocked with a protein-free solution (G2052; Servicebio) for 10 min, incubated with primary antibodies overnight at 4 °C, then washed with Tris-Buffered Saline with Tween® (25 mM Tris, 0.15 M NaCl, 0.05% Tween-20, pH 7.5; Thermo Fisher Scientific), and incubated with secondary antibodies for 1 h at room temperature. Protein bands were visualized using a General ECL Chemiluminescent Detection Kit (PK10001; Proteintech, China) and a ChemiDoc™ Touch Imaging System (Bio-Rad).
The following primary antibodies were used: anti-MMP13 (1:1000; Proteintech); anti-MMP9 (1:1000; Proteintech); anti-ADAMTS5 (1:1000; Abcam); anti-COL2A1 (1:1000; Proteintech); anti-CH25H (1:1000; Santa Cruz Biotechnology); anti-SOX9 (1:1000; Proteintech); anti-ACAN (1:1000; Proteintech); anti-CY7B1 (1:1000; Santa Cruz Biotechnology); anti-RORα (1:1000; Abcam); and anti-GAPDH (1:2000; Proteintech). All of the primary antibodies were diluted in a primary antibody dilution solution (P0023A; Beyotime). The following secondary antibodies were used as appropriate: goat anti-mouse or goat anti-rabbit (1:5000; Proteintech). The relevant information of the antibodies used is summarized in Table S5.
RNA sequencing and bioinformatic analysis
SMSCs were categorized into two groups (Ctr and LNPs@S/T) and induced for seven days. Three biological replicates from each group were analyzed. Chondrocytes were divided into two groups: IL-1β and LNPs@S/T. They were first induced with IL-1β for 6 h, and then the chondrocytes in the LNPs@S/T group were inoculated on the surface of the hydrogel for culture. Three biological replicates were analyzed in each group. Total RNA was extracted with TRIzol and sequenced by Qingdao Oebiotech Co. Ltd. The gene expression value was transformed as log10 [TPM (Transcripts Per Million reads) + 1]. The RNA sequencing data were normalized via the fragments per kilobase per million reads method. FPKM3 of each gene was calculated and the read counts of each gene were obtained by HTSeq-count4. PCA analysis were performed using R (v 3.2.0) to evaluate the biological duplication of samples. Differential expression analysis was performed using the DESeq25. Q value < 0.05 and foldchange > 2 or foldchange < 0.5 was set as the threshold for significantly differential expression gene (DEGs). Hierarchical cluster analysis of DEGs was performed using R (v 3.2.0) to demonstrate the expression pattern of genes in different groups and samples. The radar map of top 30 genes was drew to show the expression of up-regulated or down-regulated DEGs using R packet ggradar. Based on the hypergeometric distribution, KEGG pathway and GO analysis of DEGs were performed to screen the significant enriched term using R (v3.2.0).
In vivo evaluationRat model of OA
All animals were obtained from Jinan Pengyue Laboratory Animal Breeding Co. Ltd. (Jinan, China). In the sham group, eight female Sprague–Dawley rats (8 weeks old, average weight 150 g) underwent a mock surgery with their knee joints rinsed with saline and stitched without further treatment. An OA model was induced in another 32 rats by ACLT, which involved opening the knee joint, exposing, and cutting the ligament. Success was confirmed by significant tibial displacement when the knee was bent to 90 degrees. The joint was then cleaned with sterile saline and sewn shut. Four weeks later, these rats were divided into four treatment groups: PBS, S/T, LNPs, and LNPs@S/T. Both the sham and ACLT groups received their respective treatments:
Sham group: 60 μL PBS was injected intra-articularly into the rats that underwent skin incision but not anterior cruciate ligament rupture.
PBS group: 60 μL PBS was injected intra-articularly into the rats after anterior cruciate ligament rupture.
S/T group: 60 μL of peptide hydrogel was injected intra-articularly into the rats after anterior cruciate ligament rupture.
LNPs group: 60 μL LNPs were injected into the joint cavity of rats after anterior cruciate ligament rupture.
LNPs@S/T group: 60 μL of peptide hydrogel mixed with the same dose of LNPs as in the LNPs group was injected into the joint cavity of rats after anterior cruciate ligament rupture.
In vivo tracking of LNPs
Cy5-labeled siRNA-encased LNPs were incorporated into the hydrogel. To test the hydrogel's extended release, three Sprague–Dawley rats underwent ACLT and received intra-articular injections of 60 μL LNPs or LNPs@S/T. The cartilage targeting efficiency of C5-24-LNPs was assessed by comparing injections of targeted and non-targeted LNPs into rat knee joints. Imaging was performed on days 1, 3, 7, 10, and 14 (IVIS Lumina LT; Perkin Elmer, Waltham, MA).
Evaluation of LNPs@S/T biocompatibility
Blood samples (200 µL) were collected from the eye socket venous plexus at 1, 2, and 4 weeks post-injection for hematological analysis. Subsequently, the animals were euthanized using CO2 inhalation, and major organs were harvested and fixed for 24 h for histology.
Micro computed tomography (micro-CT)
High-resolution micro-CT (Quantum GX2, Japan) was utilized to acquire detailed images of rat knee joints, specifically targeting the knee cartilage, distal femur, and proximal tibia. The data were reconstructed using a micro-CT workstation. Three researchers evaluated osteochondroses and measured parameters such as bone mineral density (BMD), new bone volume (BV), and bone volume fraction (BV/TV) with CT Analyser software (version 1.11, Skyscan).
Histopathology, immunohistochemistry, and immunofluorescence
To evaluate cartilage histology, rat knee joints collected at 7 and 10 weeks were fixed in 4% paraformaldehyde, decalcified with EDTA (Servicebio, China), dehydrated in ethanol, and embedded in paraffin. The embedded paraffin blocks were cut into 3 μm thick slices using a microtome (RM2016, Shanghai Leica Instrument Co. Ltd.). Sections were stained with hematoxylin and eosin (H&E) and safranin-O/fast green. Immunohistochemical staining was used to evaluate inflammatory markers (MMP13) and chondrogenic markers (COL2A1, SOX9, ACAN) at 7 and 10 weeks after surgery. For immunofluorescence, sections were incubated with anti-CD90 primary antibodies (Abcam, Cambridge, UK) at a dilution of 1:400 and anti-CD73 (Abcam, Cambridge, UK) primary antibodies at a dilution of 1:500 overnight at 4 °C and then incubated with Alexa Fluor-conjugated secondary antibodies (Life Technologies, Waltham, MA) at a dilution of 1:500 for 1 h at room temperature. After DAPI counterstaining, sections were imaged using an automated digital slide scanner (VS200; Olympus, Tokyo, Japan).
Statistical analysis
Data are presented as means, with error bars indicating the standard deviation (SD) from independent samples. Animal groupings were randomized prior to treatment initiation. Statistical comparisons were made using one-way analysis of variance, or two-tailed Student's t-test, as specified in the figure legends. All statistical analyses were performed using Prism software (GraphPad). A P value < 0.05 was considered to indicate a statistically significant difference.







留言 (0)