Animals
Male BALB/c mice (19–21 g) were obtained from the SPF Biotechnology Co., Ltd. (Beijing, China). Mice were housed under the constant condition of humidity (50 ± 5%) and temperature (25 ± 1 °C) with 12–12 h light‒dark cycles. Food and water were available ad libitum.
Primers, bacteria and plasmids
Bacteria, plasmids, and primers used in this study are described in Table S1 and Table S2. MRAB and DRPA were used for modeling in this study. MRAB (ATCC17978) was purchased from American Type Culture Collection (ATCC, Rockefeller, USA), and DRPA was isolated from clinical samples and kindly given by Prof. Xie Fei. All bacteria were grown at 37 °C in LB broth or on LB plates solidified with 2% agar. If necessary, an appropriate antibiotic (100 µg mL−1 apramycin, or 10 µg mL−1 tetracycline) was added to the solid or liquid media to screen engineered bacteria.
CRISPR/Cas9 plasmid construction and electrotransformation
The CRISPR/Cas9 plasmids, pABK against MRAB and pPAK against DRPA, were constructed by Gibson Assembly (NEB, Ipswich, USA), and the construction details can be found in the additional file. Both pABK and pPAK can be digested by BsaI enzyme and then introduced into different sgRNA fragments through T4 ligase. Both pABKs and pPAK were verified by Sanger sequencing (GENEWIZ, Suzhou, China). Electrocompetent cells of MRAB and DRPA were prepared according to the previous reports [44, 51]. An aliquot (5 µL) of CRISPR/Cas9 plasmids was added to the electrocompetent cells for electrotransformation with 2.5 kV, 200 Ω, and 25 µF. The bacteria were recovered in the antibiotic-free LB media (1 mL) and shaken at 37 °C for 1 h before spreading on the LB plates supplemented with appropriate antibiotics. The plates were incubated at 37 °C overnight for colony imaging.
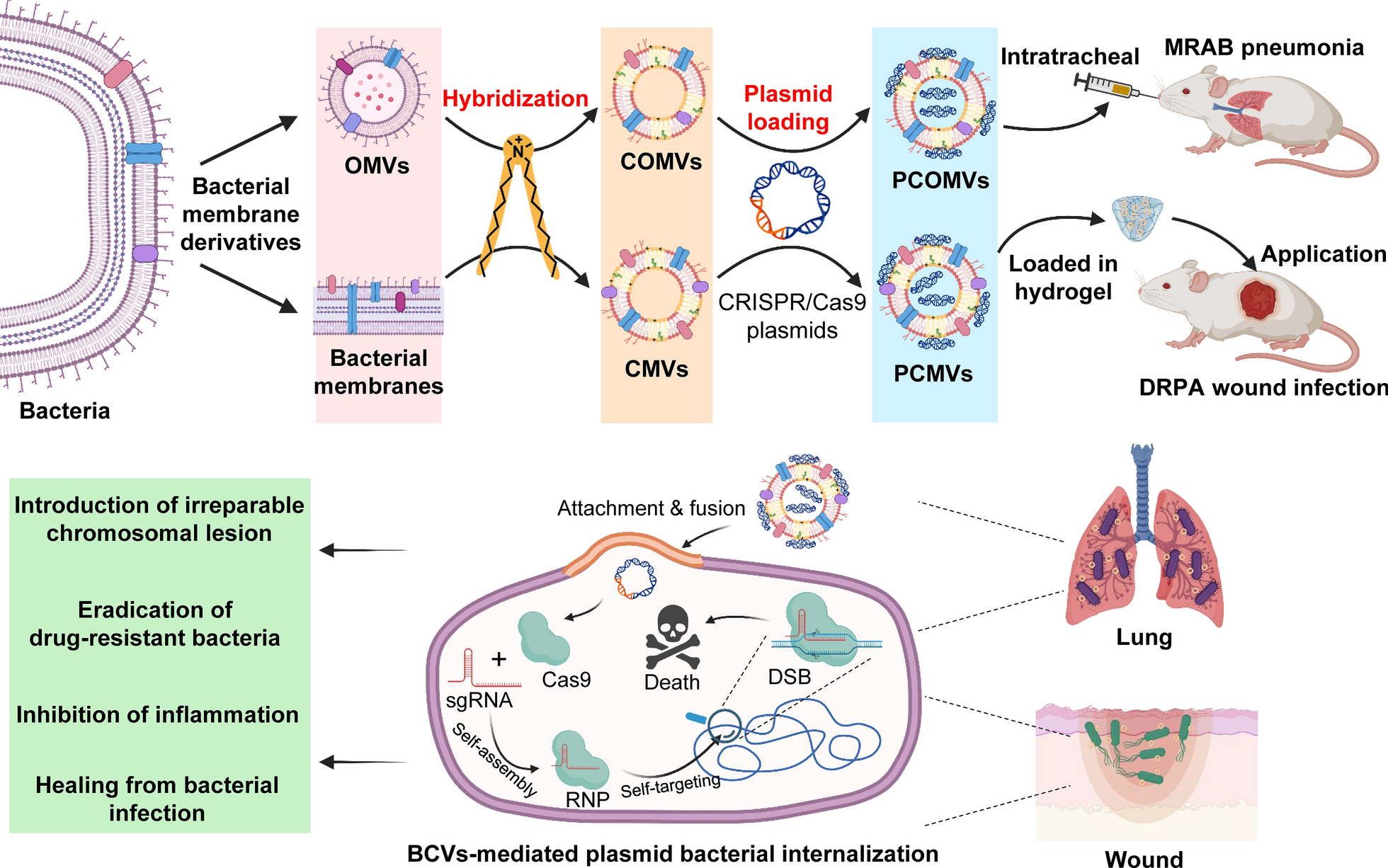
Bacterial membrane derivative isolation and formulation preparation
OMV isolation was conducted as follows. A single colony of bacteria was cultured in 200 mL of LB media at 37 °C and 220 rpm overnight until OD600 reached 1.0. The bacterial suspension was centrifuged at 5000 ×g for 15 min, and the supernatant was filtered sequentially through 0.45-µm and 0.22-µm filters (JET BIOFIL, China). The filtrate was concentrated with Millipor® centrifugal filters (MW cutoff, 100 kDa, Burlington, USA) and the concentrates underwent ultracentrifugation (Optima XPN-80, Beckman, Brea, USA) at 150,000 ×g and 4 °C for 70 min. The OMVs in the bottom were collected and washed twice with phosphate-buffered solutions (PBS). The final precipitate was suspended with PBS, wherein the proteins were determined with the BCA Protein Assay Kit and diluted to 0.25 mg mL−1 proteins in the OMVs with PBS. The OMVs were stored at −80 °C. The OMVs of MRAB harboring pABCas9 were isolated as the process, but they were named SOMVs and resuspended in LB media.
The isolation of membrane vesicles (MVs) was conducted as follows. A single colony of DRPA was cultured in LB media at 37 °C and 220 rpm until OD600 reached 0.6. The bacterial suspension was centrifuged at 5000 ×g for 15 min washed by PBS for three times (5000 ×g for 15 min), and then resuspended in PBS. The bacteria resuspension solution was ultrasound-processed using a probe ultrasound instrument (JY92-11, Ningbo Xinzhi Biotechnology Co. Ltd., Ningbo, China) with the following conditions: 300 W, ultrasound for 10 s, stop for 10 s, and repeating 100 times in an ice-cold bath, followed by 10,000 ×g centrifugation for 20 min, and the supernatant was collected and filtered through 0.22-µm filters three times. Finally, the MVs of DRPA were obtained and stored at −80 °C.
Cationic liposomes (CLs) were prepared using the film method. Briefly, DDAB (30 mg), egg yolk lecithin (EPC, 20 mg), and cholesterol (10 mg) were dissolved in absolute ethanol (6 mL) under ultrasound and transferred to a 250-mL round flask. The ethanol was removed by rotary evaporation under vacuum at 45 °C for 1 h, resulting in a film at the bottom. The film was hydrated at 45 °C with PBS (12 mL) to obtain a suspension that was ultrasound-processed using a probe ultrasound instrument with the following conditions: 300 W, ultrasound for 10 s, stop for 10 s, and repeating 45 times in an ice-cold bath. The CLs were collected after the final suspension was filtered through 0.22-µm filters.
Cationic lipid hybrid OMVs (COMVs) and cationic lipid hybrid MVs (CMVs) were prepared with the probe ultrasound method. The CLs and the OMVs (containing 0.25 mg mL−1 proteins) were mixed with the volume ratios of 1:0.25, 1:0.5, 1:0.75, 1:1, 1:1.25, and 1:1.5, respectively. The mixtures were processed using ultrasound at 300 W for 10 s, followed by a 10-s pause, and this cycle was repeated 45 times in an ice-cold bath. The resulting solution was filtered through 0.22-µm filters to obtain COMVs. The CLs and the MVs (containing 0.25 mg mL−1 proteins) were mixed with the volume ratios of 10:3 following ultrasound-processed as above and filtered through 0.22-µm filters to obtain CMVs. COMVs or CMVs (100 µL) were mixed with the CRISPR/Cas9 plasmid (5 µg) and electroporated with two pulses at 500 V, 200 Ω, and 25 µF. The mixture was 4-fold diluted with PBS. The pABK-loaded CLs, OMVs, and pPAK-loaded MVs were also prepared by the same method to generate pABK- loaded CLs (PCLs), POMVs, and PCMVs.
PCMV-loaded hydrogels were prepared for the in vivo efficacy test. PVA-1788 (4 g) was dissolved in water (40 mL) and stirred until completely dissolved, and then underwent repeated freeze-thawing to form a cross-linked structure for freeze-drying. The PCMV suspension with an adjusted concentration (4 ng µL−1 pPAK) was added to the freeze-dried PVA gel to obtain the PCMV-loaded hydrogel for wound topical application.
Agarose gel electrophoresis
Several nucleic acid samples were prepared and the details were described in the Supporting Information. All the nucleic acid samples were detected by agarose gel electrophoresis at 110 V for 30 min, and the nucleic acid was stained with Goldview® dye (Yeasen, Shanghai, China). The agarose gels were observed by an imager (OI600MF, BIO-OI, Guangzhou, China).
Western blotting and SDS-PAGE
MRAB harboring pABCas9 was cultured until OD600 reached 1, and then the culture was collected and centrifuged at 4 °C and 5000 ×g for 15 min. The cell precipitates were lysed after 100 °C heating for 10 min and proteins were determined using the BCA method. The samples were analyzed with the Western blotting (WB) method using the Cas9 antibodies (Bioss, Beijing, China). The MRAB bearing no pABCas9 was used as the control. CLs, OMVs, and COMVs were analyzed with the same WB method using Omp38 antibodies (AmyJet Scientific, Wuhan, China).
All samples were normalized to an equal protein concentration with the BCA method. An aliquot (20 µL) sample (including CLs, OMVs, COMVs, MVs, CMVs) was mixed with loading buffer and heated to 100 °C for 10 min, and then the samples were loaded onto a 10% acrylamide gel and subjected to electrophoresis at 80 V for 1 h followed by 120 V for 2 h. The gels were stained with Coomassie blue (Epizyme, Shanghai, China) for photographing.
Fluorescence detection of formulations
The fusion of CLs and OMVs was verified with the Förster resonance energy transfer (FRET) method. Briefly, dye-labeled CLs were prepared with the same method as normal CLs, except that 25 µg of NBD-PE (excitation/emission = 470/534 nm) and 36 µg of Rhod-PE (excitation/emission = 560/583 nm) were dissolved in 400 µL of the lipid mixture solution (DDAB, 2 mg; EPC, 1.33 mg; cholesterol, 0.67 mg). The molar ratio of DDAB, EPC, cholesterol, NBD-PE, and Rhod-PE was 75:74:30:0.5:0.5. The free dyes were removed by centrifugation at 16,000 ×g for 15 min for three times. The dye-labeled CLs were mixed with OMVs and processed as above. The fluorescent spectra (500─700 nm) were recorded at the excitation wavelength of 470 nm with a microplate reader (SPARK, Tecan, Männedorf, Switzerland). The fusion of CLs and the MVs derived from DRPA was verified by the same method. For visualization, DiD and DiO dyes were used to label CLs and OMVs, respectively. The two membranes were mixed and processed as above. Fluorescent images were captured using a confocal laser scanning microscope (CLSM 880, Carl Zeiss AG, Oberkochen, Germany).
Physical property determination of CLs, OMVs, COMVs, and PCOMVs
The formulations (CLs, OMVs, COMVs, PCOMVs, MVs, CMVs, and PCMVs) were stained with uranyl acetate (1%) and observed under a transmission electron microscope (TEM, H-7650, Hitachi, Tokyo, Japan), respectively. The size distribution and zeta potentials of them were measured by dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern Instruments, Malvern, UK) at 25 °C.
Co-localization of formulations and bacteria
CLs, OMVs, and COMVs were labeled with DiD (10−5 M) for 20 min, respectively, and washed with PBS thrice after centrifugation to remove excess DiD. The labeled CLs, OMVs, and COMVs were incubated with MRAB (108 CFU mL−1) at 37°C for 6 h. The MRAB was stained with 4,6′-diamidino-2-phenylindol (DAPI) (2 µg mL−1) and images were captured using Zeiss CLSM 880.
Bacterial viability staining and scanning electron microscopy
PBS, SM, COMVs, pABK, and PCOMVs were co-incubated with MRAB as described above, respectively, and then the bacteria were collected and washed thrice with PBS; the bacteria were stained with SYTO-9 (5 µM) and propidium iodide (PI, 1 µg mL−1) at 37 °C for 15 min, following observation under a microscope (CKX53, Olympus Corporation, Tokyo, Japan). The bacteria in the precipitates were fixed with glutaraldehyde, dehydrated with alcohol, coated with gold, and observed under a scanning electron microscope (SEM, S-4800, Hitachi, Tokyo, Japan) at 5 kV.
CK-8 assay
Beas-2B cells were seeded in 96-well plates at a density of 2 × 104 cells/well and incubated at 37 °C for 24 h. A series of PCOMVs in BEpiCM media with pABK concentrations ranging from 0.063 ng µL−1 to 0.250 ng µL−1 were added into the wells, respectively, and the cells were sequentially incubated at 37 °C for 24 h. The culture media containing 10% cell counting kit-8 (CCK-8) were added into each well followed by incubation for 1 h. The absorbance of wells was measured at 450 nm with a microplate reader and the cell viability was calculated as Eq. (1).
$$}\left( \% \right) = (}_}}} - }_}}} )/(}_}}} - }_}}} ) \times }00\%$$
(1)
Toxicity tests of PCOMVs
The hemolysis assays of formulations were conducted with rat red blood cells (RBCs) [62]. Briefly, a 2% RBC suspension was mixed with different concentrations of PCOMVs (3, 5, 7 ng µL−1 pABK in PCOMVs), PBS (negative control), and 1% Triton X-100 solutions (positive control) at 37 °C for 2 h, respectively. After the suspensions were centrifuged at 2000 rpm and 4 °C for 10 min in a centrifuge (Sorvall™ Legend™ Micro 21R, Thermo Scientific, Waltham, USA), the OD of supernatants was measured using a microplate reader (Spark, TECAN, Männedorf, Switzerland) at 540 nm. The hemolysis rates were calculated according to Eq. (2).
$$}\left( \% \right) = \left( }_}}} }_}}} } \right)/\left( }_}}} }_}}} } \right)} \times }00\%$$
(2)
The in vivo toxicity of inhaled PCOMVs was assessed on mice after i.t. administration of PCOMVs referred our previous study [63]. Briefly, mice were anesthetized and fixed on a mouse operating table; under a laryngoscope, PCOMVs were sprayed into the lung of mice through the trachea via a thin tube. Mice were randomly divided into 4 groups, including the healthy group without any process, the other three groups with i.t. administration of 20 µL PCOMVs containing 3, 5, and 7 ng µL−1 pABK for every mouse, respectively. After 24 h, whole blood was collected. Primary blood cells, including white blood cells (WBC), RBC, and platelets (PLT) were measured using a blood cell analyzer (DF52-Vet, Shenzhen Mindray Bio-Medical Electronics Co. Ltd.; Shenzhen, China). Glutamic-pyruvic transaminase (ALT) and creatinine (CRE) in the serum were also measured.
IVIS imaging
PCOMVs (1 mL) were mixed with a DiR solution (5 µM, 1 µL) in dimethyl sulphoxide (DMSO) at 37 °C for 0.5 h to obtain DiR-labeled PCOMVs that were i.t. administered to mice. The mice were sacrificed after 6, 12, and 24 h, respectively, and their major organs, including the heart, liver, spleen, lung, and kidney, were excised and imaged (748/780 nm) using an in vivo imaging system (IVIS) (ABL-X5 Plus, Tanon, Shanghai, China).
Pneumonia model and treatment
Mouse bacterial pneumonia model was established as referred to our previous research [64]. Briefly, a dexamethasone acetate injection (5 mg mL−1, 0.1 mL) was intraperitoneally (i.p.) injected into mice for 5 consecutive days (from day − 4 to day 0), and then anesthetized with isoflurane. MRAB (25 µL, 106 CFU mL−1) was i.t. administered to the mice. Six hours later, the drug formulations (20 µL) were sprayed into the lung through the trachea with an insufflator (Model DP-4, Penn-Century, Inc., USA).
The mice were randomly divided in 5 groups with 5 mice in every group. The formulations included PBS, SM (32 µg mL−1), COMVs, pABK (1 ng µL−1), and PCOMVs (1 ng µL−1 pABK). The healthy mice in the healthy group were only treated with i.t. PBS as above. The body weight of mice during the experiment was recorded. On day 4, the blood oxygen saturation (SpO2) was examined with pulse oximeter (HS20A, Heal Force Bio-Meditech Holdings Limited, Shanghai, China). Their behavioral open-field test was performed to assess the exercise capacity of mice with each trial lasting 5 min. The total distance covered by each mouse was recorded. A meticulous cleaning of the apparatus was carried out between animals using a 20% (v/v) ethanol solution. The upper lobes of the right lung were processed as above. Tissues were deparaffinized and rehydrated, followed by the standard terminal-deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) staining and microscopic examination. Their pulmonary histopathology was conducted as above described.
Wound infection model and treatment
Mouse bacterial wound infection models were established as referred to our previous reports [11, 65]. The mice were anesthetized with i.p. injection of 1% pentobarbital sodium (50 mg kg−1), and their dorsum hair was removed. The mice were scalded at 100 °C for 6 s using an instrument (YLS-5Q, Bio-will Co., Ltd., Shanghai, China) to form a circular, deep, partial-thickness burns wound with the diameter of ~ 1 cm on the mucous layer. A DRPA suspension (6 × 108 CFU mL−1, 20 µL) in PBS was evenly dripped onto the wound, and Tegaderm™ Film (1624 W, 3 M, Saint Paul, USA) was applied to the surface of the skin until yellow-green pus was produced.
The mice were randomly divided into 4 groups, including the uninfected control, infected control, kanamycin-loaded PVA hydrogel (Kan, 0.5 mg kg−1), and PCMV-loaded PVA hydrogel (PCMV, pPAK 0.4 ng µL−1) groups (n = 6). The wounds in the 2 treatment groups were covered with the hydrogels of 0.2 mL, respectively. Both Kan and PCMVs were refreshed on day 3 and 7. The wounds were photographed on day 0, 3, 7, 14, and 21, and the wound area was analyzed using ImageJ software (the National Institute of Health, USA). Wound recovery rates were calculated as Eq. (3). A0 represents the wound area on day 0, and At indicates the wound area at measuring time points. All the mice were sacrificed on day 21, and the wound tissues were excised. The skin samples of 4 mice each group were used to determine inflammatory cytokines, and the other samples were used in histopathological study as previous described.
$$}\left( \% \right) = (A_ - A_ )/A_ \times }00\%$$
(3)
Inflammatory cytokine assays
The upper lobes of the left lung or the skin tissues were homogenized with sterile saline to get 10% (w/w) homogenates. The homogenates were centrifuged at 5000 ×g for 10 min at 4 °C, and the supernatant was collected for TNF-α and IL-6 detection following the ELISA kit operating manuals (Enzyme-linked Biotechnology Co., Ltd., Shanghai, China). OD450 was determined using a microplate reader (Spark, TECAN, Männedorf, Switzerland).
Data analysis
All data were presented as mean ± standard deviation (SD). The statistical analysis was performed with GraphPad Prism (Version 8.0, GraphPad Software, San Diego, USA). One-way analysis of variance (ANOVA) was employed to identify significant differences between different groups. Significance levels were denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.







留言 (0)