
記住我
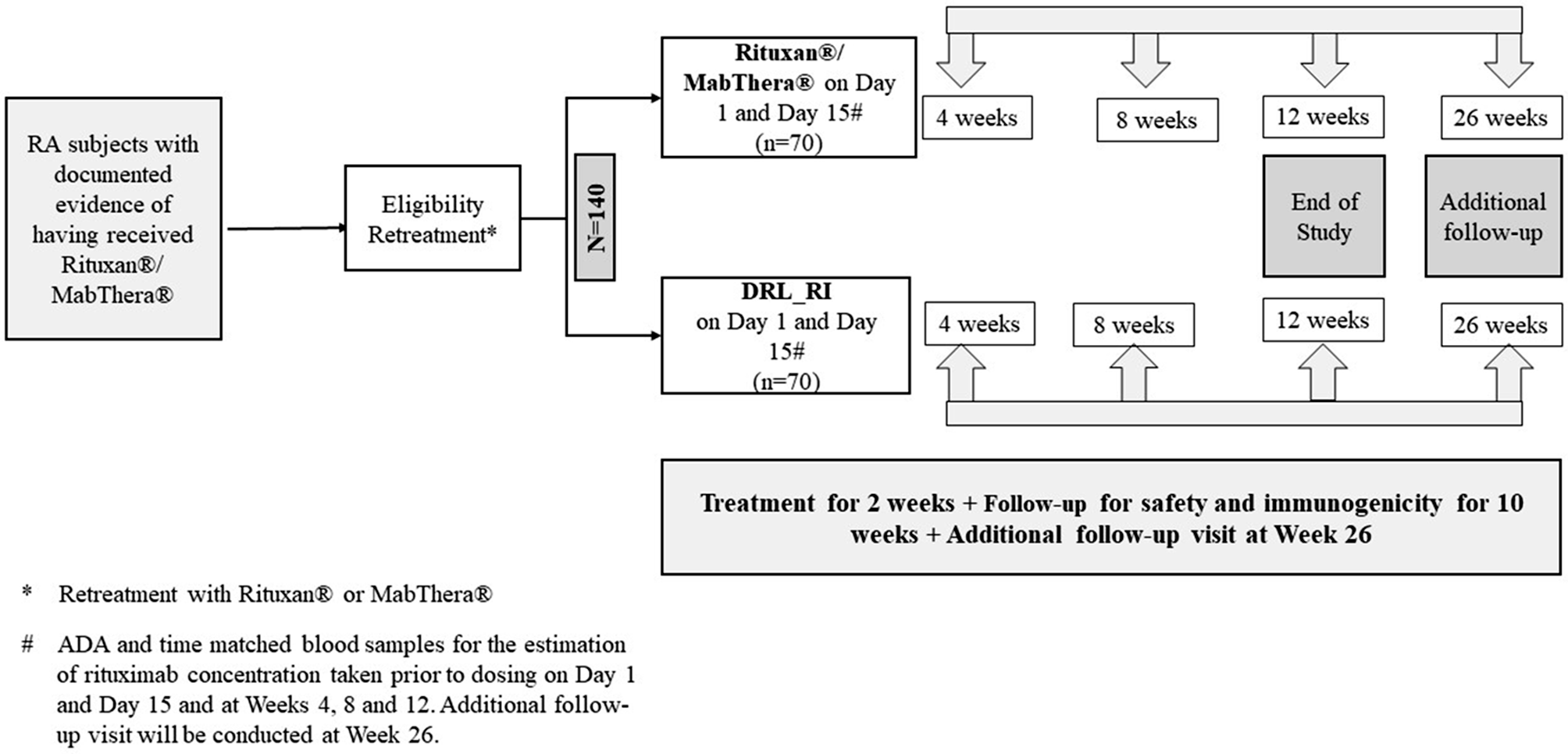




A total of 224 patients were screened, of which, 140 patients were randomized to receive DRL_RI (n = 70) or RP/RMP [RP for those already on RP (n = 22) and RMP for those already on RMP (n = 48) before study entry]. Data for RP/RMP was pooled into one group for comparison with the DRL_RI group. Of the enrolled 140 patients, 138 (98.6%) patients completed study treatment; 2 (2.9%) patients were discontinued from DRL_RI group– one due to an AE, and one due to consent withdrawal. In all, 134 (95.7%) patients completed the study till Week 12, and 118 patients re-consented for Week 26 follow-up visit. Of these, 116 (98.3%) patients completed Week 26 follow-up: 57 [96.6%] patients from DRL_RI group and 59 [100%] patients from RP/RMP group. Treatment compliance and trial discontinuation between groups were similar. All 140 enrolled patients (70 patients from each group) were included in Safety Population. Immunogenicity and TMRC populations included 137 (97.9%) patients: 69 [98.6%] from DRL_RI and 68 [97.1%] from RP/RMP groups (CONSORT Flow Chart Fig. 2). COVID-19 related protocol deviations occurred in 18 (12.9%) patients; the most frequent significant deviation was missing endpoint assessments in 4 (5.7%) patients of DRL_RI group and 5 (7.1%) patients of RP/RMP group.
Fig. 2
CONSORT Flow Chart for Patient Disposition. Abbreviations: ADA, anti-drug antibody; DRL_RI, biosimilar rituximab; f/u, follow-up; N/n, number of patients; TMRC, time-matched rituximab concentration
Patient demographics and baseline characteristics were comparable between groups (Table 1). The mean (SD) age of patients was 59.8 (11.7) years [range: 24, 86 years] and mean (SD) body-mass index (BMI) was 27.8 (6.2) kg/m2. The majority of patients were female (82.1%), postmenopausal (71.1%), White (99.3%), and ‘Not Hispanic or Latino’ (88.6%). Most patients (67.9%) were recruited from Europe. In all, 122 (87.1%) patients—63 (90.0%) patients in DRL_RI group and 59 (84.3%) patients in RP/RMP group—reported at least one medical condition/surgery at baseline; hypertension, osteoarthritis, and osteoporosis were common. The median duration of RA at randomization was 112 months, and median time from prior rituximab treatment was 6.6 months (mean: 7.6 months). Overall, 45 (32.1%) patients had prior exposure to RP and 95 (67.9%) patients to RMP. Fewer patients [(47 (33.6%)] received one prior course of rituximab and majority of patients [93 (66.4%)] received more than one prior course of rituximab.
Table 1 Baseline demographics and Disease characteristics (all enrolled patients)All 140 patients received Day 1 dose, and 137 patients received Day 15 dose — 69 patients received DRL_RI, 22 patients received RP and 46 patients received RMP. Day 1 dose was interrupted in 1 patient in DRL_RI group due to IRR, and in 1 patient in RP/RMP group due to hypersensitivity. Common concomitant medications were folic acid (97.1% in both groups), MTX (100% in both groups), and cholecalciferol (34.3% in DRL_RI and 22.9% in RP/RMP group). About 41% patients in each group received systemic glucocorticoids. One patient in DRL_RI group received methylprednisolone and one patient in RP/RMP group received triamcinolone as a rescue medication. One (1.4%) patient from DRL_RI group received re-dosing of rituximab (Truxima®, a rituximab biosimilar) at the end of the study visit post completion of the safety follow-up in the study (Week 29) based on the investigator’s discretion.
Immunogenicity resultsThree (4.3%) patients in DRL_RI group were ADA positive at baseline (pre-dose); all tested ADA- negative after dosing and none were NAb positive. Post dosing with DRL_RI, 1 (1.4%) new patient tested ADA positive on Day 15, Week 8, and Week 12, was also NAb positive at Week 8 but later tested NAb negative at Week 12. Titres for these ADA-positive patients decreased by the end of the study (Table 2). One (1.5%) patient in RP/RMP group tested ADA positive at baseline (pre-dose). This patient and another patient tested ADA positive at Week 12; none of these two patients were NAb positive till Week 12, though the titres did not decrease in these patients. Overall, ADA and NAb incidences were comparable with no significant differences in the ADA titres between the two groups in the study (Table 2).
Table 2 Summary of Antidrug Antibody Evaluations in study patients (Immunogenicity Population)Time-matched rituximab concentration (TMRC)Median TMRC values were comparable between the groups: 88.63 µg/mL in DRL_RI group and 100.8 µg/mL in RP/RMP group on Day 15. Week 4 showed the highest median TMRC: 141.2 µg/mL for DRL_RI vs. 159.4 µg/mL for RP/RMP. At Week 8, the median TMRC declined to 49.1 µg/mL and 69.9 µg/mL, respectively, and further to 20.3 µg/mL and 29.9 µg/mL, respectively, at Week 12. Blood levels in the treatment arms were comparable and did not show interference in the detection of immunogenicity.
Safety resultsTEAE incidence was comparable between DRL_RI (34.3%) and RP/RMP groups (38.6%). Overall, the incidences of drug-related TEAEs, TEAEs leading to treatment discontinuation, Grade ≥ 3 TEAEs, and treatment-emergent SAEs were not different between the groups. Two (2.9%) patients in DRL_RI group reported drug- related IRR Grade 1—itching in the throat and roof of the mouth in 1 patient, and nausea after infusion in other patient—none were serious nor led to any treatment discontinuation. No anaphylactic reactions were reported (Table 3).
Table 3 Summary of adverse events (Safety Population)Common TEAEs in RP/RMP vs. DRL_RI group included infections and infestations (18.6% vs. 8.6%), gastrointestinal disorders (8.6% vs. 4.3%), nervous system disorders (8.6% vs. 1.4%), and musculoskeletal and connective tissue disorders (4.3% vs. 2.9%). Common adverse events (> 3%) in RP/RMP group included diarrhoea and headache (7.1% of patients, each) and COVID-19 and nasopharyngitis (4.3% of patients, each); no AEs were reported in > 3% frequency in DRL_RI group.
Drug-related TEAEs included IRRs (2.9%) and diarrhoea (1.4%) in DRL_RI group (overall 4.3%), while dizziness, embolic stroke, headache, bronchitis, pharyngitis, hypersensitivity, and rash, each in 1.4% patients in RP/RMP group (overall 5.7%). Grade 3 TEAEs in DRL_RI group included COVID-19 pneumonia, myocardial infarction, and bile duct stone in 1 patient each. Grade 4 TEAE of fungal infection and a Grade 5 TEAE of COVID-19 pneumonia, both, were reported in 1 patient; Grade 5 COVID-19 event was fatal. Grade 3 TEAEs reported in RP/RMP group were cystitis in 1 patient; and empyema, septic shock, enteritis, and embolic stroke in 1 patient. No Grade 4 or Grade 5 TEAE was reported in RP/RMP group.
Four (5.7%) patients in DRL_RI group and 2 (2.9%) patients in RP/RMP group experienced an SAE. Treatment-emergent SAEs in DRL_RI group included COVID-19 pneumonia in 2 patients resulting in death of 1 patient, myocardial infarction and intestinal resection in 1 patient, each; none of these were related to DRL_RI. Treatment-emergent SAEs in RP/RMP group included enteritis, cystitis, empyema, septic shock, and embolic stroke, each in 1 (1.4%) patient; of these, embolic stroke was related to RP/RMP.
DRL_RI was discontinued in 1 patient due to Grade 2 drug hypersensitivity (to amlodipine), not related to DRL_RI; while RP/RMP was discontinued in 1 patient due to Grade 2 hypersensitivity considered related to rituximab. The incidence of EOSI (COVID-19 and related) was not relevantly different between DRL_RI (3 patients) and RP/RMP groups (4 patients). In DRL_RI group, 1 patient had Grade 2 COVID-19, and 2 patients had serious COVID-19 pneumonia (Grade 3 and Grade 5-fatal). In RP/RMP group, 1 patient had Grade 2 COVID-19 pneumonia and 3 patients had Grade 2 COVID-19, which resolved during the study.
留言 (0)