
記住我

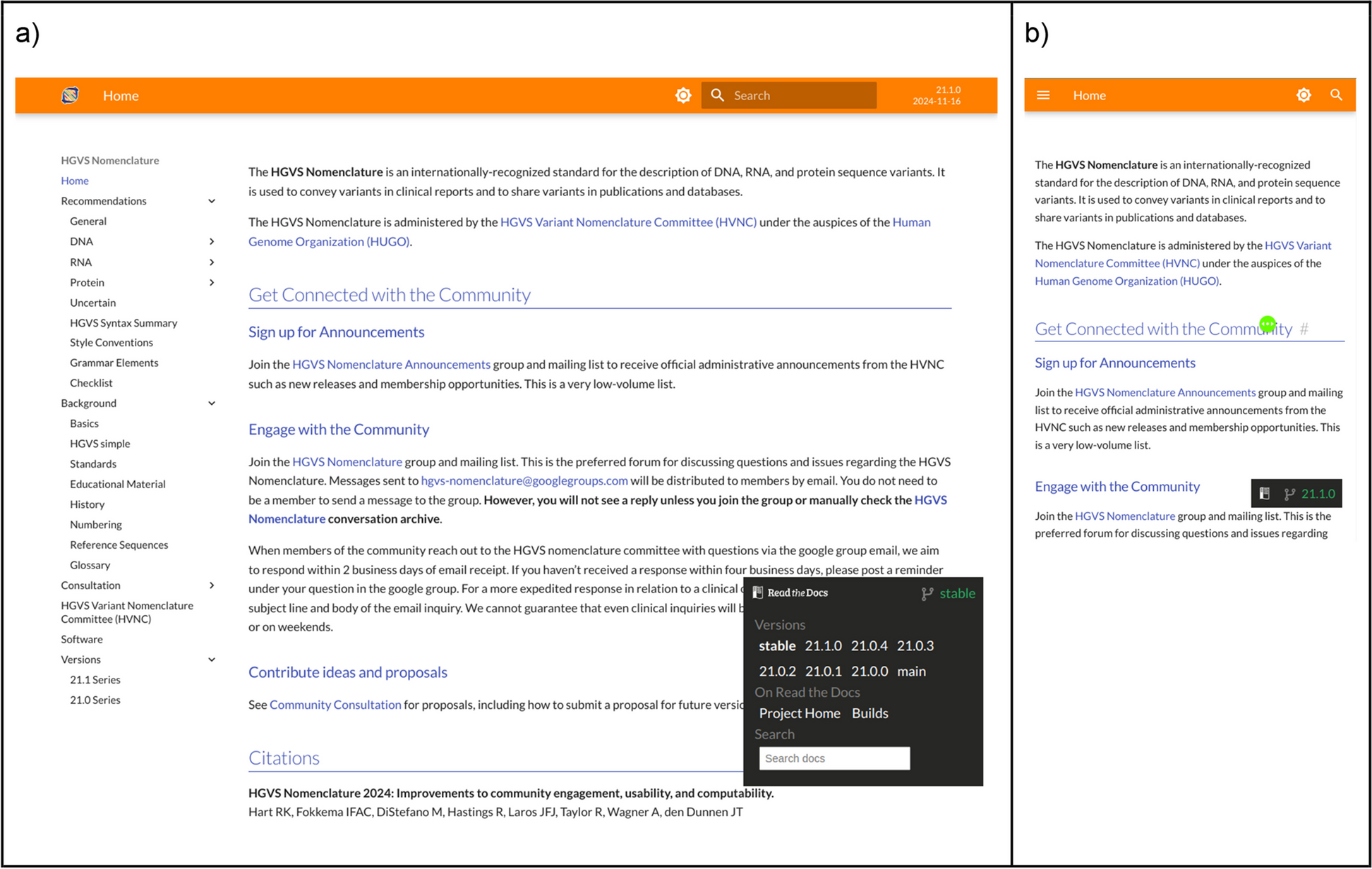


The previous version of the HGVS Nomenclature website was written using a mix of Markdown [16] and the HyperText Markup Language (HTML), which made it difficult to update and format consistently. Although changes were listed on a specific Versions page, the previous site presented only one version of the HGVS Nomenclature, making it difficult to investigate the evolution of the recommendations. The new site is written nearly entirely using Markdown for consistency, rendered using MkDocs [17], and automatically deployed to Read the Docs [18].
Many of the most significant new features of the HGVS Nomenclature website were created based on feedback from users. Examples include the collapsible navigation that makes it easy to understand the site layout, the version number shown in the top panel on all pages (Fig. 1), and a new versions page that itemizes changes between versions (Fig. 2). The new website enables users to easily switch between versions of the HGVS Nomenclature. Other major changes to the website include corrections of numerous small errors and inconsistencies, integrated searching with previews, the ability to navigate between different versions of the HGVS Nomenclature, and consistent styling that provides for distinguishing valid examples and invalid counterexamples (in red). The new website is responsive and remains functional across a wide range of devices, including phones, tablets, and laptops. An archived, prior version of the HGVS Nomenclature website (version 20.05) remains available [19].
Fig. 1
a The landing page for hgvs-nomenclature.org shows the “foldable” navigation menu on the left, a search bar with a clear indication of the selected version, and the Read the Docs version selector to navigate to other versions. The version selector is minimized and unobtrusive by default but expanded here to show functionality. b Mobile version of the landing page. The navigation menu, search features, and selected version are hidden in the “hamburger menu” denoted by the ≡ symbol
Fig. 2
The release notes page provides details of every change to the website since the previous version
Change processes, versioning, and release processesThe processes for updating the HGVS Nomenclature now follow conventional source code management practices. The GitHub repository [20], which stores the official documentation and code of the HVNC, is managed by a group of maintainers. Significant changes are discussed prior to implementation, and all changes are submitted as “pull requests” for review by maintainers prior to incorporation. As appropriate, the maintainers will release the aggregated changes with a new version tag and release notes. The site will be built and deployed automatically, typically within a few minutes.
The HNVC has developed a new versioning policy based on “semantic versioning” [21] that is intended to help users and adopters manage compatibility between providers and consumers of tools. Each release of the HGVS Nomenclature will be tagged with a version that has the format X.Y.Z, often called the major, minor, and patch numbers, respectively. When backward incompatible changes are made, the major version (X) will be incremented; such changes are expected to be very rare. When new features or capabilities are added, the minor version (Y) will be incremented. For trivial changes, such as typographic fixes or clarifications, the patch version (Z) will be incremented. Because the patch level carries no change in the intent of the HGVS Nomenclature, two versions may be compared by the major and minor versions alone.
The HGVS Nomenclature now recommends that data providers — that is, tools or databases that present HGVS variant descriptions — should advertise the version of the HGVS Nomenclature that they use. Similarly, data consumers — that is, websites or software tools that accept variant descriptions as input — should advertise the versions of the HGVS Nomenclature that they accept. Data consumers should use the same major version as the provider and a minor version that is greater than or equal to that of the provider; other combinations risk backward incompatibility (when the consumer’s major version is less than the provider’s major version) or lack of a feature required to parse a variant (when the consumer’s minor version is less than the provider’s minor version). Details are given on the HGVS Nomenclature Versions page [22].
Presentation of HGVS nomenclature grammarHistorically, the HGVS Nomenclature was presented as a set of guidelines and an informal grammar primarily aimed at human readers, providing recommendations for describing sequence variants. This approach was accessible to readers but required human interpretation and preference that is no longer suitable when building interoperable and scalable systems for the analysis of sequence variation. To enhance the precision, consistency, and interoperability of sequence variant descriptions, the HGVS Nomenclature is transitioning to the use of Extended Backus-Naur Form (EBNF) for defining its syntax [23]. EBNF is a formal notation that uses symbols and patterns to rigorously define the structure of a language, making it both transparent to human readers and precise enough for automated processing by software. This transition is essential for improving the reliability of sequence variant descriptions in computational systems, as it enables the development of tools that can automatically validate, parse, and interpret these descriptions with minimal ambiguity. By adopting EBNF, the HGVS Nomenclature is better positioned to support scalable genetic analyses, ensuring that descriptions are not only consistent across different platforms but also interoperable with other genomic standards and tools.
HGVS Nomenclature grammar is stored in a single, computer-readable file structure indexed by molecule type (DNA, RNA, protein) and variant type (substitution, deletion, etc.). All presentations of HGVS Nomenclature syntax rules are generated from this single file. Because the current HGVS Nomenclature is not easily represented as a strictly valid grammar, this release adopts the syntax of EBNF for variant descriptions without attempting to reconcile missing or ambiguous elements of the previous release. Nonetheless, the new presentation often clarifies rules that were previously difficult to interpret unambiguously. For example, the HGVS Nomenclature allows two distinct forms of DNA substitutions: a “simple” substitution on a reference sequence and a more complex form that imposes a transcript structure on an underlying genomic sequence. The existence of two forms is not obvious on the previous site but is clearly presented in the new website (Fig. 3).
Fig. 3
a Syntax for a DNA substitution from the previous website [24]. b Syntax for a DNA substitution from the updated website [25]. The new presentation clarifies that there are two distinct forms of a DNA substitution variant and includes an explanation of the syntactic components of each form
The HGVS Nomenclature has historically been grouped by molecule type and then classified by variant type. This arrangement makes it difficult for users to identify commonalities of a variant type across molecule types. For example, it is difficult to understand how DNA, RNA, and protein forms of substitutions might appear together and users often needed to navigate at least three pages to identify the rules for a single variant type, leading to frequent requests for reorganization. Storing grammar rules in a single file enables reuse and reformatting to summarize the syntax rules by variant type, thereby facilitating comparisons across molecule types (Fig. 4).
Fig. 4
An excerpt of the new HGVS Syntax Summary pages [26]. The HGVS Syntax Summary shows all syntax rules in HGVS, organized primarily by variant type and secondarily by molecule type. This organization complements that of the website and facilitates readers to compare syntax rules. The syntax rules in the summary and molecule-specific page (Fig. 3b) are drawn from the same file, thereby guaranteeing consistency between these two views
List of available softwareAs the HGVS Nomenclature migrates towards the full adoption of a computational grammar, software will become essential to using the standard reliably and effectively. The new website includes a list of software that has been submitted by the authors and deemed to meet reasonably objective criteria [27]. Software must be a library, web API, or web user interface. Only openly available software will be listed, and well-recognized licenses by the Open Source Initiative are preferred. Currently, software may claim one or more of a set of functionality categories in order to help users find tools appropriate for their uses. Tools must also be primarily intended to manipulate HGVS expressions; tools that merely use HGVS expressions for input or output are not included. Tools that have tests and/or were described in a peer-reviewed publication are also preferred. To submit a tool to be included in the software list, developers can submit a pull request to the HGVS Nomenclature repository [20].
New and updated features in the HGVS nomenclature specificationWhile most of the recent changes to the HGVS Nomenclature focused on how the specification is maintained and communicated, two substantive changes were made in response to timely requests from the community. A new syntax for gene fusions enables the HGVS Nomenclature to be used for adjoined transcripts. In addition, the HGVS Nomenclature 21.0 updates guidance for the selection of transcripts.
Aligning conventions for adjoined transcripts and gene fusionsAs defined by VICC, a gene fusion is “the joining of two or more genes resulting in a chimeric transcript and/or a novel interaction between a rearranged regulatory element with the expression of a partner gene transcript” [28, 29]. An adjoined transcript results from the transcription of a gene fusion and is represented in HGVS using a double colon, e.g., NM_002354.2:r.−358_555::NM_000251.2:r.212_*279. This definition, alongside the definitions of chimeric transcript fusions and regulatory fusions, has been added to the HGVS Nomenclature glossary. Prior definitions of the terms “RNA Fusion” and “Fusion Transcript” (from community proposal SVD-WG007) have been deprecated in favor of the community-aligned term “adjoined transcript,” focusing on the precise semantics of representing two adjoined transcript sequences. A new page describing the HGVS syntax for adjoined transcripts is available under the RNA Recommendations section of the HGVS Nomenclature.
The VICC fusion nomenclature is compatible with the HGVS Nomenclature recommendations for intervening nucleic acid sequences (described as linker sequences) and adjoined transcripts. It also provides recommendations for an exon-based nomenclature that shares the structure of adjoined transcripts as used in the HGVS Nomenclature. In recognizing that the human genomics community has a need for representing regulatory fusions, exon-based chimeric transcript fusions, and gene-level fusions[29], the HGVS Nomenclature and ISCN now endorse the use of the VICC nomenclature for the representation of gene fusion events, in addition to their prior recommendation to use the HGNC naming conventions [30] for fusion genes.
Updated guidance for selection of transcriptsHistorically, the HGVS Nomenclature has recommended the use of the longest transcript of a gene that represents the known biological and clinical significance of that locus. However, due to transcript sequence differences between RefSeq and Ensembl, and differences between transcripts and the primary assembly, the choice of transcript was practically challenging.
The Matched Annotation from the NCBI and EMBL-EBI Select (MANE) transcript set defines representative transcripts and corresponding proteins for human protein-coding genes [31]. MANE transcripts share identical exon structure and sequence between NCBI and Ensembl, and therefore, identifiers from those databases may be used interchangeably. Furthermore, MANE transcript sequences match the GRCh38 reference assembly identically, which avoids complications that arise with variation that overlaps alignment mismatches and gaps between transcripts and GRCh37. The MANE Select subset defines one transcript at each protein-coding locus that represents the known biology at that locus. The HGVS Nomenclature now recommends the use of MANE Select and MANE Plus Clinical transcripts where possible and appropriate. In addition, the previous recommendation to use Locus Reference Genomic sequences was withdrawn.
留言 (0)