
記住我



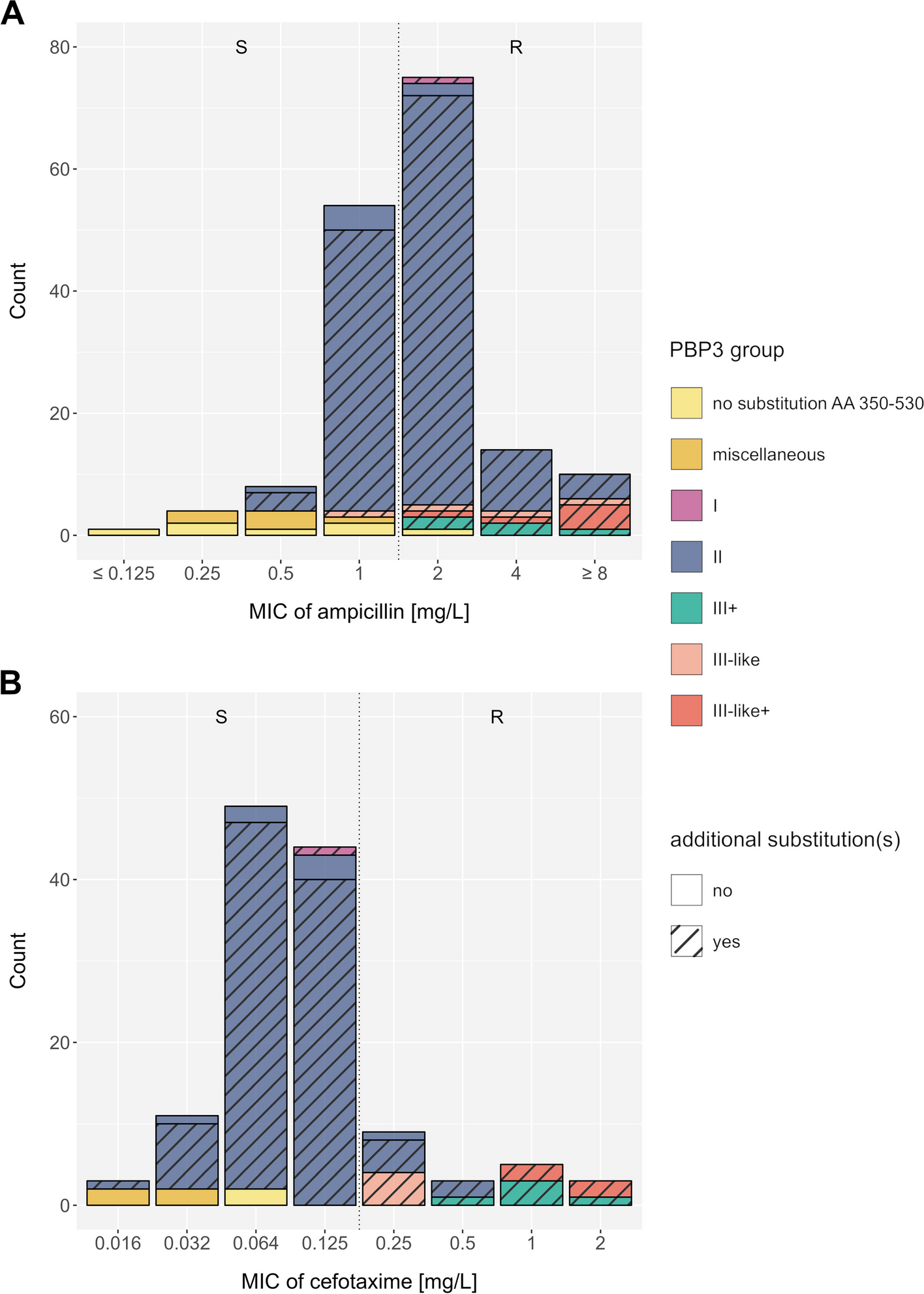
We performed a systematic literature review and meta-analysis to correlate ampicillin and cefotaxime resistance with existing PBP3 group definitions. Overall, 46 studies were eligible for screening the full text, of which 34 (74%) did not meet the eligibility criteria, mainly due to the presentation of collated/pooled data only, or the absence of DNA sequencing data. One additional study was excluded because of nomenclature conflicts of reported mutations (Additional file 1: Fig. S2, Additional file 2: Table S1).
In total, we included genotypic and phenotypic data from 12 studies on 291 β-lactamase negative H. influenzae isolates in the meta-analysis. Quantitative drug susceptibility testing (MIC determination) was performed by gradient diffusion strips (e.g., Etest by bioMérieux/AB Biodisk, E-strip by Oxoid) and interpreted using EUCAST clinical breakpoints (n = 167 isolates; 57.47%) as well as broth microdilution (BMD) with CLSI recommended medium and clinical breakpoints (n = 124 isolates; 42.6%) and BMD with EUCAST recommended medium and clinical breakpoints (n = 63 isolates; 21.6%), respectively. Ampicillin MIC values determined by at least one of the above methods were available for 284/291 isolates (97.6%), and cefotaxime MIC values were available for 134/291 isolates (46.0%). Of those 146/284 (51.4%) were resistant to ampicillin according to EUCAST (or at least intermediate according to CLSI) (MIC > 1 mg/L). Concerning cefotaxime resistance, 20/127 (15.7%) isolates tested with gradient diffusion strips and broth microdilution using EUCAST recommended media were resistant (MIC > 0.125 mg/L), whereas all 7 isolates tested with broth microdilution using CLSI recommended media and clinical breakpoints were susceptible (MIC ≤ 2 µg/mL). Data for other β-lactams were scarce and therefore not used for further analysis.
For each isolate, we converted the reported mutation pattern into a PBP3 group according to the consensus nomenclature proposed by Nürnberg et al. (Table 1) [23]; other nomenclatures that have previously been used are listed in Additional file 2: Table S4. The distribution of PBP3 groups was as follows: group I (n = 6), group II (n = 235), group III + (n = 13), group III-like (n = 8), and group III-like+ (n = 8), while no isolate belonged to group III. Most of the isolates classified into the resistance groups also harbored additional mutations within the transpeptidase domain (Fig. 1, Additional file 1: Fig. S5 and S6). The vast majority of H. influenzae isolates lacking any reported PBP3 substitution in the analyzed transpeptidase domain, or not matching any of the proposed PBP3 groups were ampicillin (19/21) and cefotaxime susceptible (6/6) (Table 1, Additional file 2: Table S5). All PBP3 groups and subgroups with sufficient data (n > 5) were significantly associated with ampicillin resistance (p < 0.05), except group IIa, which was borderline significant (p = 0.06) (Table 1). For group III sensu stricto and group I, we could only identify < 6 isolates, and statistical significance was not assessed. For cefotaxime, only group III-related isolates were significantly associated with cefotaxime resistance (p = 0.02, Additional file 2: Table S5); individual subgroups could not be assessed due to the low sample size. For both ampicillin and cefotaxime, the reported associations remained robust for MICs determined with gradient diffusion test only (data not shown). Overall, MIC differences and categorical classifications into resistant and susceptible between gradient diffusion test and broth microdilution were only deemed marginally different for our cohorts (Additional file 1: Fig. S7). Notably, many group II isolates were still tested phenotypically susceptible to ampicillin with both, gradient diffusion test and broth microdilution (Fig. 1, Additional file 1: Fig. S5, S6, and S7), and thus specificities to predict resistance were low (15.6%) (Table 1). Further stratification within group II could also not resolve these discordances (Additional file 1: Fig. S8). In fact, the EUCAST breakpoint for ampicillin at 1 mg/L divides group II isolates into a resistant and a susceptible population (Fig. 1A). With regard to cefotaxime, the EUCAST breakpoint at 0.125 mg/L classified the majority of group II isolates, tested using gradient diffusion strips (100/110, 90.9%), as susceptible (Fig. 1B). Individual mutations alone were poor predictors of an ampicillin- and cefotaxime-resistant phenotype, most of which occurred in both susceptible and resistant isolates. The PBP3 substitution L389F was always related with phenotypic resistance to ampicillin and cefotaxime in tests interpreted with EUCAST breakpoints (Additional file 1: Fig. S9A-D).
Table 1 Associations of previously proposed PBP3 groups and ampicillin resistance for 276 β-lactamase negative H. influenzae isolates with stated ampicillin MIC valuesFig. 1
Distribution of minimum inhibitory concentrations (based on gradient diffusion strips (e.g., Etest)) for A ampicillin (n = 167) and B cefotaxime (n = 127) among previously published β-lactamase negative H. influenzae isolates. The color code represents the main PBP3 groups, isolates with no reported substitutions within the PBP3 transpeptidase domain AA 350–530 (compared to the reference strain Rd KW20), and isolates with miscellaneous PBP3 transpeptidase substitution patterns (not matching the minimum set of mutations used to describe known groups). Hatched bars display isolates exhibiting additional PBP3 substitutions at position 350 to 530, other than the respective group-specific substitutions. Dotted vertical lines indicate clinical breakpoints (EUCAST) that differentiate phenotypically susceptible (S) and phenotypically resistant (R) isolates
Population genomic analysis of PBP3 mutations and groups in a global H. influenzae cohortNext, we sought to investigate whether some of the previously proposed PBP3 resistance-associated groups might be phylogenetically informative, i.e., whether isolates belonging to the same PBP3 group also cluster together in a global phylogeny. To this end, we used a population genomic approach with a global collection of 555 H. influenzae isolates deposited in the pubMLST database [26]. The collection included a diverse non-redundant set of isolates from 21 countries (1944–2022) (see “ Methods”). The isolates belong to 414 different STs and represent 287 different HAEM1263 alleles (loci in pubMLST corresponding to full-length ftsI) and 158 ftsI alleles (loci in pubMLST corresponding to the PBP3 transpeptidase domain (AA 326–532)) (Additional file 2: Table S2).
Overall, 149/555 isolates (26.8%) were classified into one of the previously described resistance groups, 394/555 isolates (71.0%) had none of the group-specific substitutions, and 12/555 isolates (2.1%) had either only A502T, A502S, or A502V, respectively (Additional file 2: Table S2). No group III representative substitution pattern, sensu stricto, was identified. Interestingly, neither of the serotype a, d, or c isolates had group-specific mutations. Out of 149 isolates classified in one of the resistance groups, 42 (28.2%) had additional group-specific mutations aside from the minimum set defining the group (Table 1). For instance, within group IIb (A502V + N526K), the majority (35/53, 66%) also had the additional substitution M377I, which is characteristic of group III strains. The global phylogeny of H. influenzae (Fig. 2) shows that group-specific resistance mutations and combinations are not phylogenetically informative as they are distributed across independent lineages (i.e., sequence types).
Fig. 2
Phylogeny of a global and diverse collection of 555 H. influenzae isolates based on a core-genome alignment of 1429 genes. The category “other” represents all genomes without a PBP3 group-specific substitution or with a PBP3 substitution at position 502 only. Branches that have a local support value of less than 0.8 are highlighted with red dots. AAS amino acid substitutions in PBP3, NTHi non-typeable H. influenzae
Interestingly, N526K and R517H rarely occurred together in one isolate (Fig. 3). On the other hand, we confirmed the genomic linkage of group III mutations, i.e., L389F, S385T, and M377I. Specifically, the mutation L389F always co-occurred with the group mutations M377I and S385T in our global dataset (Fig. 3), and L389F mostly co-occurred with D350N and S357N, two other putative resistance-related substitutions (Additional file 2: Tables S2 and S4). Overall, the pairwise comparison of all group-specific substitutions revealed very common substitution patterns as well as combinations that never co-existed in one isolate (Fig. 3).
Fig. 3
Co-occurrence of PBP3 substitutions in a global H. influenzae collection (n = 555). Hierarchical clustering based on proportions of PBP3 group-specific substitutions on the y-axis that co-occurred with PBP3 substitutions on the x-axis
Next, we performed a homoplasy analysis of the full ftsI gene in order to identify additional mutations possibly under positive selection. Notably, this analysis might also highlight alleles under relaxed purifying selection that have no measurable effect on the bacterial fitness. Overall, we identified 272 mutations showing signs of homoplasy, i.e., identical mutations in phylogenetically unrelated subgroups. Plotting the top 100 hits from the homoplasy analysis, as judged by the lowest consistency indices, suggested an accumulation of mutations within the transpeptidase domain of the ftsI gene (Additional file 1: Fig. S10). However, we also identified numerous other mutations outside this domain in the homoplasy analysis and likely neutral synonymous mutations not affecting the protein structure (Additional file 2: Table S6). The top PBP3 substitutions from the homoplasy analysis were V547I, E603D, D350N, N569S, and P31S, respectively.
Genome-wide variant associations with ampicillin resistance in a new clinical H. influenzae cohortIn order to identify additional ftsI mutations and genes other than ftsI that might be related to ampicillin resistance and to better explain the observed MIC distributions (Additional file 1: Fig. S11), we performed two microbial genome-wide association studies (GWAS) employing a novel clinical cohort. First, we used log2-transformed MIC values as phenotype (available for 247 isolates). Second, we used the binary phenotypic classification (available for 298 isolates), i.e., resistant or susceptible based on EUCAST guidelines, in the following referred to as “resistance status”. In both approaches, the only prominently associated locus was ftsI (Fig. 4A and B). In the GWAS using the binary classification resistant or susceptible, several ftsI variants had p-values of less than 10–14, whereas all other genomic variants had p-values greater than 10–9 (Fig. 4B). Likewise, in the GWAS based on MIC values, ftsI variants had association p-values less than 10–25 in comparison to p-values larger than 10–17 for other genome-wide variants (Fig. 4A). The most strongly associated PBP3 substitution in both GWAS approaches was M377I (p = 2.49 × 10–14 (MIC); p = 4.36x10−15 (resistance status)). We identified another four non-synonymous PBP3 substitutions that were more strongly associated with resistance than all other substitutions (p-value ≤ 10–8, Additional file 2: Table S7), namely the PBP3 group substitutions A502V and N526K as well as V547I and N569S (located outside the transpeptidase domain), in addition to several synonymous ftsI variants. These five are also the most strongly MIC-associated substitutions (Additional file 2: Table S7).
Fig. 4
The -1 × log10 transformed p-values of variant associations genome-wide. In panels A and B, each dot represents a variant, in panels C and D, each dot represents a gene. A Manhattan plot of MIC GWAS. B Manhattan plot of resistance status GWAS. C Gene-wise most significant MIC-association p-value (x-axis) versus most significant resistance status-associated p-value (y-axis). Whereas C considers all variants occurring in more than 10 isolates, D considers thereof only the subset of variants causing amino acid substitutions. Only ftsI had in both GWASs a much smaller p-value compared to the remaining genes. Respective HTML-based interactive figures providing detailed information on individual data points at mouse-over are provided as Additional file 3: Material S3 (panel A), 4 (panel B), 5, (panel C), and 6 (panel D)
Next, we investigated a possible role of other genes in ampicillin resistance, by selecting the most strongly associated variant per gene from both GWAS approaches and contrasting the respective p-values (Fig. 4C and D)).
Except for ftsI, genes were preferentially associated with ampicillin resistance in only one of the two GWASs. Thus, no additional strongly supported candidate resistance gene could be identified. However, an interesting gene that came up consistently with top amino acid substitution p-values in both GWASs was a yet uncharacterized protein encoded by Rd_05960 (NCBI locus tag RdKW20_001113) (Fig. 4A and D). In addition, genes ridA and ompP2 rank comparably high in both GWASs. The gene usg encoding an oxidoreductase is the second most strongly associated gene in MIC GWAS (Fig. 4A and C) and gene ettA in resistance status GWAS (Fig. 4B and C), both, however, based on the presence of synonymous mutations that are typically considered non-functional. Additional genes with amino acid substitutions that are strongly associated preferentially in one of both GWASs comprise prmB, tauA, oppA, and frmA (GWAS based on resistance status, Fig. 4B and D) as well as metE, hcpC, msbB, and ftsP (GWAS based on MIC values, Fig. 4A and D). Finally, the intergenic variant most strongly associated with resistance status is located approximately 400 bases upstream of hslR, which encodes for a heat shock protein 15 homolog (Fig. 4B and C). The intergenic variant most strongly associated with MIC values is located within a cluster of molybdate ABC transportation molecules modA, modB, modC, and modE, upstream of modB, modC, and modE (Fig. 4A and C).
Fine-mapping of the ftsI locusWe subsequently aimed to investigate how well resistance is explained by ftsI mutations. To this end, we used 2 joint models of 44 high-quality non-synonymous mutations in ftsI detected in a minimum of 10 isolates within our novel clinical cohort, 1 for resistance status, and 1 for MIC values (Additional file 2: Table S7). Surprisingly, the joint model also poorly predicted resistance status, and likewise, MIC values were poorly modeled by ftsI mutations (model-adjusted R2 = 0.5603). Only a few mutations significantly contributed to resistance phenotypes, nine with respect to MIC values and seven with respect to resistance status, with A502V (p = 0.01385 (MIC), p = 0.02010 (R/S)), N526K (p = 0.00816 (MIC), p = 0.06775 (R/S)), and V547I (p = 0.00456 (MIC), p = 0.00784 (R/S)), the only common significant substitutions in both models (significance level 0.1, Additional file 2: Table S7). The significant mutations from both joint models included not only the top substitutions from our GWASs, i.e., M377I, A502, V547I, N526K, and N569S, but also substitutions that are not linked to resistance when considered individually, E603D, A586S, E135Q, and N75S. Notably, N569S and V547I are in high linkage disequilibrium, a measure of nonrandom co-occurrence of alleles at two sites (squared Pearson correlation coefficient r2 = 0.71, Additional file 1: Fig. S12 and interactive HTML-based Additional file 3: Material S7) as well as the combination of M377I, A502V, and N526K (A502V/N526K r2 = 0.87; M377I/N526K r2 = 0.48; M377I/A502V r2 = 0.41, Additional file 1: Fig. S12). This was also apparent when these mutations were plotted on a phylogenetic tree: the combinations of the substitutions M377I, A502V, and N526K (i.e., group IIb-defining mutations + M377I) always co-occurred with V547I, N569S as well as with D350N/S (Additional file 1: Fig. S13). N569S always co-occurred with V547I but not vice versa. Ampicillin MICs of group IIb isolates harboring the combination M377I, A502V, and N526K (n = 58) were slightly higher than those for group IIb isolates without M377I (n = 28 with MIC available) (median 1.5 vs 1 mg/L, p = 0.005, Mann–Whitney U test). Likewise, MICs for cefotaxime were increased for PBP3 group II isolates with PBP M377I (n = 37) compared to group II isolates lacking the PBP M377I substitution (n = 18) (median 0.064 vs 0.047 mg/L, p = 0.028, Mann–Whitney U test).
Mutations in ftsI resulting in PBP3 group substitutions L389F and R517H were identified in less than 10 isolates of the clinical cohort and thus were not assessed via GWAS and joint model. The PBP3 group substitution I449V was not associated with resistance throughout, and S385T was only weakly associated with resistance in the MIC GWAS and joint model (p = 0.03210, Additional file 2: Table S7).
Finally, we investigated in the clinical cohort whether additional ftsI mutations or combinations thereof stratify previously established resistance groups and allow an improved prediction of phenotypic ampicillin resistance. Towards this, we constructed haplotypes representing all observed combinations composed of 44 ftsI mutations that are present in a minimum of 10 isolates in the clinical cohort and that cause PBP3 substitutions (12 within and 32 outside the transpeptidase domain) (Fig. 5 and Additional file 1: Fig. S14). Overall, the observed combinations resulted in 83 haplotypes, 11 of which represented more than 50% of the isolates (Additional file 2: Table S8). The most common haplotype in the clinical cohort (here denoted H1 with n = 24 isolates), found in both Portugal and Germany (cf. Additional file 1: Fig. S14), carried the previously mentioned combination of PBP3 substitutions M377I, A502V, and N526K, with ampicillin MICs greater than or equal to 1 mg/L (isolates from ST 1034 (n = 11), 14 (n = 12), and 1206 (n = 1); Additional file 2: Table S3). This combination was also present in other common haplotypes (H6 with 11 isolates from ST 367 (n = 8), 142 (n = 1), 203 (n = 1), and 136 (n = 1); H11 with 5 isolates from ST 834 (n = 3), 422 (n = 1), and 1881 (n = 1); H12 with 5 isolates from ST 203). In the entire cohort, the combination M377I, A502V, and N526K was found in 60 isolates, with only 3 isolates harboring an ampicillin MIC < 1 mg/L (Suppl. Table S3). Other haplotypes comprising mainly isolates with a MIC less than or equal to 1, i.e., susceptible isolates, are located in the center of the network from which different paths along similar haplotypes exist towards haplotypes observed mainly in resistant isolates (Fig. 5B)). Haplotypes of isolates without any PBP3 transpeptidase domain substitution as well as haplotypes of isolates that carry transpeptidase domain substitutions that do not belong to a currently defined PBP3 group are largely phenotypically susceptibility (Fig. 5). Specifically, out of 131 isolates without group-specific mutations or without any non-synonymous mutations between AA 350–530, 127 (96.9%) were susceptible. The four resistant ones (all with MIC = 4) belong to serotype a, serotype b, serotype f, and NTHi, respectively, and three carry a V547I mutation (Additional file 1: Fig. S14 and Additional file 2: Table S3).
Fig. 5
Haplotype network for 44 ftsI variants detected in a minimum of 10 out of overall 247 isolates. Nodes represent haplotypes, i.e., combinations of variants, and lines connect similar haplotypes. Nodes are scaled according to isolate frequency of the corresponding haplotype and haplotypes are named in the order of occurrence, starting with H1 as the most common haplotype. A Colors denote resistance groups and the abundant combination of group IIb variants with the additional PBP substitution M377I. B Colors denote minimum inhibitory concentrations (MIC) for ampicillin. Blue shades relate to isolates with MIC values considered ampicillin susceptible (MIC < 1 mg/L); red shades indicate isolates classified as ampicillin resistant (MIC > 1 mg/L). Borderline MIC values of 1 mg/L (by definition ampicillin susceptible) are indicated in gray
留言 (0)