
記住我
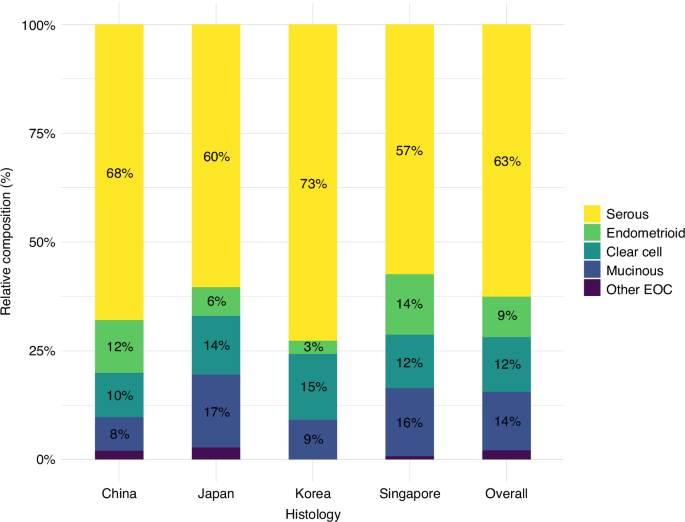
Firstly, we evaluated the expression level of PTGDS based on data from Oncomine database, and also found the higher expression of PTGDS in PTCL in comparison with normal T cells (p < 0.001) (Fig. 1a). Then, analysis based on GSE6338 database verified the high expression of PTGDS in PTCL patients (Supplementary Fig. S1A–C). Moreover, results from IHC assay showed that the expression level of PTGDS protein was higher in PTCL tissue (n = 159) than that in control samples (n = 38) (Fig. 1b, c). Similarly, compared with normal T cells from PBMCs of healthy donors, the expression of PTGDS was increased in PTCL cell lines (Fig. 1d). As PTGDS was a kind of secreted protein, we further evaluated the level of serum PTGDS and found that compared with healthy control (n = 31), PTCL patients (n = 76) displayed lower level of serum PTGDS (p < 0.01) (Supplementary Fig. S1D), indicating the reduced secretion of PTGDS protein in PTCL cells.
Fig. 1: High expression of PTGDS was associated with tumor progression of PTCL.
a The expression of PTGDS mRNA in tissue from PTCL patients was higher than that in normal T cells based on Oncomine database. b Representative pictures of immunohistochemical staining from PTCL tissue and control sample. Bar = 40 μm. c Statistic analysis showed the increased expression of PTGDS protein in PTCL tissue (n = 159) in comparison with control samples (n = 38). d Results of western blotting showed that the expression level of PTGDS protein was higher in PTCL cell lines than that in normal T cells. e IHC score of PTGDS was found to be associated with clinical features in PTCL patients. f, g Kaplan–Meier survival analysis revealed that the positive expression of PTGDS was associated with worse OS and PFS in PTCL patients. Data are shown as the mean ± SD. ***p < 0.001.
To investigate the clinical association of PTGDS in PTCL patients, we performed further analysis and found that the high expression of PTGDS in tumor tissue was statistically correlated with ALK+ ALCL (p = 0.008) and elevated ESR (p = 0.004) in PTCL patients (Fig. 1e, Supplementary Fig. S1E). However, there was no significant difference between PTGDS expression and Ann Arbor stage, IPI socre, liver invasion and so on (Supplementary Table 1), which might due to the limited number of samples in different subtypes. Moreover, the high level of serum PTGDS ( > 56.9 ng/mL) was found to be associated with clinical characteristics in PTCL patients, including high IPI score, elevated β2-MG, advanced stage and elevated ESR (Supplementary Fig. S1F, Supplementary Table 3). Furthermore, Kaplan–Meier survival curve showed that PTCL patients with positive expression of PTGDS exhibited shorter overall survival (OS, p = 0.001) and progression-free survival (PFS, p = 0.003) (Fig. 1f, g). Besides, univariate and multivariate analyses revealed that PTGDS score was independent risk factor for OS and PFS in PTCL patients (Supplementary Tables 5 and 6). These results provided a promising biomarker for the prognostic prediction of PTCL patients, and indicated the potential role of PTGDS in the progression of PTCL.
Knockdown of PTGDS expression inhibited tumor growth of PTCL cells in vitro and in vivoThe above findings prompted us to further illuminate the regulatory role of PTGDS in the progression of PTCL, and we performed lentiviral transfection to overexpress and knockdown the expression of PTGDS in PTCL cells. Western blotting was performed to evaluate the transfection efficiency (Fig. 2a). Results of CCK8 assay showed that PTGDS overexpression increased the proliferation of PTCL cells while PTGDS knockdown significantly decreased it (Fig. 2b). Then, sh-PTGDS#2 was chosen for further research based on the efficacy of gene knockdown and the level of proliferation inhibition (Fig. 2a, b). Moreover, rescue experiments revealed that PTGDS overexpression could partly reverse the inhibitory effects of PTGDS knockdown on cell proliferation in PTCL (Supplementary Fig. S2A), confirming the key role of PTGDS in PTCL progression. Besides, PTGDS knockdown was found to suppress the viability of PTCL cells (Fig. 2c) and the expression of c-myc, an essential factor in cell proliferation (Fig. 2d).
Fig. 2: Knockdown of PTGDS expression inhibited tumor growth of PTCL cells in vitro and in vivo.
a Western blotting was performed to verify the transfection efficiency. b Results of CCK8 assay at 48 h showed that PTGDS overexpression increased the proliferation of PTCL cells and PTGDS knockdown decreased it. c, d PTGDS knockdown inhibited the cell viability and the expression of c-myc in PTCL cells at 36 h. e–g Compared with sh-Control group, the growth rate, weight, volume and bioluminescence of tumor in mouse model were lower in sh-PTGDS group (n = 5 per group). h–j PTGDS knockdown induced cell cycle arrest at the G0/G1 phase, decreased the expression of CDK4, and increased the expression of CDK inhibitors P21 and P27 in PTCL cells at 36 h. k–m PTCL cells with PTGDS knockdown displayed higher apoptosis rate, increased expression of Bax and the cleaved forms of caspase-3, caspase-9 and PARP at 36 h. n–o PTGDS knockdown inhibited cell invasion and the expression of zeb1 and vimentin in PTCL cells at 36 h. p PTGDS knockdown regulated the expression of important proteins in tumor tissue from mouse model. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
To verify the role of targeting PTGDS in vivo, we established a subcutaneous xenograft mouse model using PTCL cells with PTGDS or control knockdown (n = 5 per group). Decreased growth rate (Fig. 2e), low tumor volume and weight (Fig. 2f, Supplementary Fig. S2B) were observed in mice bearing PTCL cells with PTGDS knockdown at the end of experiment. Furthermore, results from in-vivo imaging system showed that targeted inhibition of PTGDS could significantly decrease the bioluminescence of tumor cells (Fig. 2g). Overall, the above results indicated that targeting PTGDS could inhibit the proliferation of PTCL cells in vitro and in vivo.
To investigate the role of PTGDS in cell cycle and apoptosis, we performed flow cytometry experiment in PTCL cells with PTGDS or control knockdown. Representative pictures of cell cycle analysis were shown in Fig. 2h, and PTGDS knockdown was observed to induce cell cycle arrest at the G0/G1 phase (p < 0.05) (Fig. 2i). Moreover, targeted inhibition of PTGDS could decrease the expression of CDK4, an essential factor in the transformation from G1 phase to S phase, and significantly increase the expression of CDK inhibitors P21 and P27 in PTCL cells (Fig. 2j). Furthermore, PTCL cells with PTGDS knockdown displayed higher apoptosis rate (Fig. 2k, l), increased expression of pro-apoptotic proteins, including Bax (Bcl-2 associated X protein) and the cleaved forms of caspase-3, caspase-9 and PARP (Fig. 2m). Rescue experiments found that the promoting effects of PTGDS knockdown on cell apoptosis could be partly reversed by PTGDS overexpression (Supplementary Fig. S2C), verifying the important role of PTGDS in PTCL progression. Besides, PTGDS knockdown was found to significantly decrease the invasive ability (Fig. 2n), and the expression of zeb1 and vimentin, important positive factors of cell invasion (Fig. 2o) in PTCL cells. Similarly, tumor tissue from mice bearing sh-PTGDS cells displayed decreased expression level of c-myc, CDK4, vimentin and Ki67, and increased expression level of Bax and cleaved PARP (Fig. 2p, Supplementary Fig. S2D). Collectively, these results suggested that PTGDS might play an important role in the development of PTCL through regulating cell viability, proliferation, cell cycle, apoptosis and invasion.
PTGDS inhibitor AT56 exerted significant anti-tumor effects in PTCLTo illuminate the effects of PTGDS inhibitor AT56 in PTCL development, we performed RNA-seq in PTCL cells treated with AT56 or control, and identified differentially expressed genes (Fold change > 1.5, p < 0.05). Gene ontology (GO) analysis based on differentially expressed genes revealed that targeting inhibition of PTGDS was closely associated with tumor biology in PTCL cells, including cell death, cell apoptosis, cell cycle, cell aging, autophagy and so on (Fig. 3a).
Fig. 3: PTGDS inhibitor AT56 exerted significant anti-tumor effects in PTCL.
a RNA-seq and GO analysis showed the association between PTGDS expression and tumor biology in PTCL. b, c AT56 inhibited the proliferation and viability of PTCL cells in a dose-dependent manner at 48 h and 72 h. d AT56 decreased the expression of c-myc in PTCL cells at 36 h. e, f PTCL cells treated with AT56 for 36 h displayed dose-dependently cell cycle arrest at G0/G1 phase, the decreased expression of CDK4 and Cyclin D1 and increased expression of P21. g–i AT56 treatment for 36 h significantly increased the apoptosis rate and the expression of apoptosis-associated proteins in PTCL cells, with concentration dependence. j, k Inhibited cell invasion and decreased expression of zeb1 and vimentin were found in PTCL cells treated with AT56 for 36 h. l AT56 treatment regulated the expression of important proteins in tumor tissue from mouse model. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we performed further experiments to validate the effects of AT56 in the progression of PTCL. It’s found that AT56 could inhibit the cell proliferation and cell viability of PTCL cells, with concentration dependence (Fig. 3b, c). Besides, AT56 treatment was found to inhibit cell proliferation in sh-Con cells, but not in sh-PTGDS cells (Supplementary Fig. S3A), supporting the specificity of AT56 on PTGDS in PTCL cells. The expression of c-myc was dose-dependently decreased by the treatment with AT56 in PTCL cells (Fig. 3d), indicating the inhibitory effects of AT56 in cell growth in vitro. Moreover, AT56 was found to induce cell cycle arrest at G0/G1 phase in a dose-dependent manner (Fig. 3e). After the treatment with AT56, PTCL cells displayed decreased expression of CDK4 and Cyclin D1 and increased expression of P21 in a dose-dependent manner (Fig. 3f). AT56 treatment could also increase the early apoptotic cell populations of PTCL cells, with concentration dependence (Fig. 3g, h). Obviously, PTCL cells treated with AT56 displayed dose-dependently increased expression of pro-apoptotic proteins, including Bax, and the cleaved forms of caspase-3, caspase-9 and PARP (Fig. 3i). Moreover, apoptosis inhibitor, Z-VAD, was found to partly rescue the anti-proliferation effects of AT56 treatment in PTCL cells, but the difference was not statistically significant (Supplementary Fig. S3B), suggesting that cell apoptosis might be the important but not the major mode of cell death after targeting PTGDS. The dose-dependent reduction of cell invasion was observed in PTCL cells treated with AT56 (Fig. 3j), which was accompanied by the decreased expression of zeb1 and vimentin (Fig. 3k). Similarly, tumor tissue from mice treated with AT56 displayed decreased expression of c-myc, CDK4 and vimentin, and increased expression of cleaved PARP and Bax (Fig. 3l). Taken together, our results indicated that AT56 exerted significant anti-tumor effects in PTCL through regulating cell proliferation, cell viability, cell cycle, cell apoptosis and cell invasion.
AT56 treatment promoted the ferroptosis process in PTCL cellsTo further explore the regulatory mechanism of AT56 treatment on PTCL progression, we performed TMT-mass spectrometry in PTCL cells treated with AT56 or control. It was found that AT56 treatment could significantly regulate the expression level of ferroptosis-associated proteins in PTCL cells (Fig. 4a). Venn diagram of the overlap between up- and down-regulated ferroptosis-associated molecules in RNA-seq and TMT-mass spectrometry was shown in Supplementary Fig. S4A. KEGG pathway analysis based on the differentially expressed proteins (Fold change > 1.5, p < 0.05) in TMT mass spectrometry revealed that AT56 treatment played important role in regulating ferroptosis process in PTCL cells (Supplementary Fig. S4B).
Fig. 4: AT56 treatment promoted the ferroptosis process in PTCL cells.
a Analysis of differentially expressed proteins based on TMT-mass spectrometry showed the potential regulatory role of AT56 treatment on ferroptosis in PTCL. b AT56 treatment for 36 h increased the expression of PTGS2 in PTCL cells, classic biomarker of ferroptosis. c AT56 enhanced the inhibitory effect of Erastin and Sorafenib on cell proliferation at 36 h. d, e AT56 treatment for 36 h significantly promoted the Erastin- and Sorafenib-induced accumulation of lipid ROS. f–h Fer-1 treatment for 36 h partly reversed AT56-induced cell proliferation inhibition and lipid ROS accumulation in PTCL cells. i–k The combination of AT56 and Sorafenib significantly inhibited tumor growth in PTCL mouse model. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we further determined the regulatory role of AT56 on ferroptosis process in PTCL, and found that AT56 treatment could increase the mRNA expression of PTGS2, the hallmarks of ferroptosis, in PTCL cells (Fig. 4b). As Erastin and Sorafenib have been identified as classic ferroptosis inducers, their inhibitory effects on cell proliferation were significantly enhanced by AT56 treatment in PTCL cells (Fig. 4c). Besides, as Sorafenib functions as both ferroptosis inducer and multi-target kinase inhibitor, we found that the treatment with ferrostatin-1 (Fer-1), a specific inhibitor of ferroptosis, could reverse the proliferation inhibitory effects of Sorafenib, and the combined anti-tumor effects of Sorafenib and AT56 could be reversed by Fer-1 and DFO (Supplementary Fig. S4C), indicating that ferroptosis was the major downstream mechanism. Similarly, results of flow cytometry showed that AT56 treatment promoted the Erastin- and Sorafenib-induced accumulation of lipid ROS, a classic biomarker of ferroptosis (Fig. 4d, e). Notably, the treatment with Fer-1 reversed the inhibition of cell proliferation and the accumulation of lipid ROS in PTCL cells treated with AT56 (Fig. 4f–h), indicating that AT56 treatment might inhibit tumor growth through inducing ferroptosis process in PTCL cells.
To further verify the regulatory effects of AT56 on ferroptosis process in vivo, we constructed PTCL mouse models and found that AT56 treatment could enhance the anti-tumor effects of Sorafenib. Compared with mice received AT56 alone, Sorafenib alone and control, mice received the combination treatment of AT56 and Sorafenib displayed significantly decreased tumor growth rate (Fig. 4I), tumor volume and weight at the end of the experiment (Fig. 4j, k). Moreover, AT56 enhanced the inhibitory role of Sorafenib on the bioluminescence and Ki67 expression of PTCL cells in vivo (Supplementary Fig. S4D–E). Besides, the combination of AT56 and Sorafenib obviously regulated the expression of phenotype-related proteins, including c-myc, CDK4, cleaved PARP, Bax and vimentin (Supplementary Fig. S4F). Altogether, our in-vitro and in-vivo results indicated that AT56 treatment could promote the ferroptosis process and then inhibit tumor growth in PTCL.
Targeting PTGDS promoted the ferroptosis process in PTCL cellsTo decipher the regulatory role of targeting PTGDS on ferroptosis in PTCL, we detected ferroptosis-associated phenotypes in PTCL cells with PTGDS knockdown or overexpression. Similarly, PTGDS knockdown increased the expression of PTGS2 mRNA and enhanced the promoting effects of Erastin and Sorafenib on lipid ROS accumulation (Fig. 5a–c). Besides, the inhibitory effects of Erastin and Sorafenib on cell proliferation were significantly enhanced by PTGDS knockdown in PTCL cells (Fig. 5d). We also found that the treatment with Fer-1 and DFO could reverse the combined anti-tumor effects of Sorafenib and PTGDS knockdown in PTCL cells (Supplementary Fig. S5), indicating that ferroptosis played important role in the inhibitory effects. Notably, Fer-1 treatment could partly reverse the proliferation inhibition induced by PTGDS knockdown in PTCL cells (Fig. 5e).
Fig. 5: Targeting PTGDS promoted the ferroptosis process in PTCL cells.
a The expression of PTGS2 mRNA was increased in PTCL cells with PTGDS knockdown at 36 h. b, c PTGDS knockdown enhanced the promoting effect of Erastin and Sorafenib on lipid ROS accumulation at 36 h. d PTGDS knockdown promoted the inhibitory effect of Erastin and Sorafenib on cell proliferation at 36 h. e Fer-1 treatment reversed the proliferation inhibition induced by PTGDS knockdown at 36 h. f PTGDS overexpression decreased the expression of PTGS2 mRNA at 48 h. g–i PTGDS overexpression partly reversed the effect of Erastin and Sorafenib on lipid ROS accumulation and proliferation inhibition in PTCL cells at 48 h. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Furthermore, PTCL cells with PTGDS overexpression displayed decreased expression of PTGS2 mRNA (Fig. 5f). PTGDS overexpression was found to reverse the regulatory effects of Erastin and Sorafenib on the accumulation of lipid ROS (Fig. 5g, h) and cell proliferation (Fig. 5i) in PTCL cells. Taken together, the above results indicated that targeting PTGDS might inhibit the development of PTCL through promoting ferroptosis process.
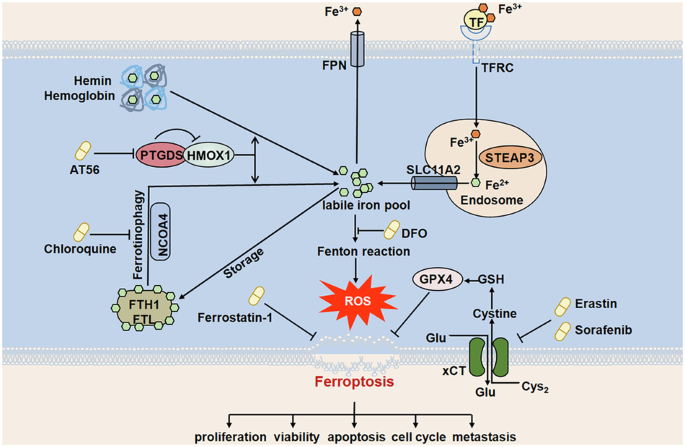
Targeting PTGDS promoted ferroptosis process through regulating iron metabolism in PTCL cellsThe major mechanisms of ferroptosis include iron accumulation, ROS production and lipid peroxidation, which are regulated by a complex regulatory network [4, 8]. As targeting PTGDS was found to promote ferroptosis process in PTCL cells, we further performed experiments to explore underlying molecular mechanism. Results of western blotting showed that AT56 treatment decreased the expression of ferroptosis-associated transcription factor NRF2, increased the expression of KEAP1, the negative regulator of NRF2, in PTCL cells in a dose-dependent manner (Fig. 6a). Similar results were found in PTCL cells with PTGDS knockdown (Supplementary Fig. S6A), indicating the enhanced ferroptosis process. Mechanically, the expression of ACSL4, the key factor in lipid metabolism, was decreased in PTCL cells with AT56 treatment and PTGDS knockdown (Fig. 6a, Supplementary Fig. S6A), indicating that targeting PTGDS promoted ferroptosis not through inducing lipid metabolism. Besides, both AT56 treatment and PTGDS knockdown increased the expression of key antioxidant regulators, xCT and GPX4 (Fig. 6a), indicating the compensatory activation of antioxidant system in ferroptosis process.
Fig. 6: Targeting PTGDS promoted ferroptosis process through regulating iron metabolism in PTCL cells.
a Western blotting results showed the expression level of ferroptosis-associated proteins in PTCL cells treated with AT56 at 36 h. b AT56 treatment for 36 h increased the level of Fe2+ in PTCL cells. c, d The increased accumulation of lipid ROS caused by AT56 was partly reversed by DFO in PTCL cells at 36 h. e DFO treatment for 48 h partly reversed the proliferation inhibitory role of AT56 in PTCL cells. f The AT56-induced accumulation of Fe2+ was reversed by DFO treatment for 36 h in PTCL cells. g The level of Fe2+ was higher in tumor tissue from mice receiving AT56 and Sorafenib. h Western blotting results showed the expression level of ferroptosis-associated proteins in tumor tissue from mouse model. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Furthermore, PTCL cells with AT56 treatment and PTGDS knockdown displayed significantly increased expression of NCOA4, FTH1 and FTL, which were important factors involved in iron metabolism [4]. Both AT56 treatment and PTGDS knockdown could increase the accumulation of bioactive Fe2+ in PTCL cells (Fig. 6b, Supplementary Fig. S6B), indicating that targeting PTGDS might promote ferroptosis process through regulating iron metabolism in PTCL.
Moreover, iron chelator DFO was found to reverse the proliferation inhibitory and lipid ROS promoting effects of AT56 treatment in PTCL cells (Fig. 6c–e). Similarly, PTCL cells with PTGDS knockdown displayed higher level of cell proliferation after the treatment with DFO (Supplementary Fig. S6C). The increased level of bioactive Fe2+ caused by AT56 treatment and PTGDS knockdown was reversed by DFO treatment in PTCL cells (Fig. 6f, Supplementary Fig. S6D), indicating the key role of iron metabolism in ferroptosis induced by targeting PTGDS in PTCL.
The regulatory role of PTGDS on iron metabolism in PTCL was further confirmed in vivo using a xenograft mouse model. Compared with mice receiving control treatment, tumor tissue from mice with AT56 treatment and PTGDS knockdown displayed increased level of Fe2+ (Fig. 6g, Supplementary Fig. S6E). Moreover, targeting PTGDS increased the expression of KEAP1, NCOA4, FTH1, FTL, and decreased the expression of NRF2 (Fig. 6h, Supplementary Fig. S6F) in vivo, which were consistent with the in-vitro results. Taken together, above results indicated that targeting PTGDS might promote ferroptosis process and tumor development through regulating iron metabolism in PTCL.
Targeting PTGDS promoted iron accumulation and ferroptosis partly through inducing ferritin autophagy in PTCLTo further explore the underlying mechanism of PTGDS on ferroptosis and iron metabolism in PTCL, we deeply analyzed the data from TMT-mass spectrometry and RNA-seq, and revealed the potential regulatory role of PTGDS on the expression of autophagy-associated molecules (Fig. 7a). As autophagy has been demonstrated to play key roles in the initiation of ferroptosis [8], we performed further experiments to illuminate the regulatory relationship between autophagy and ferroptosis when targeting PTGDS in PTCL cells.
Fig. 7: Targeting PTGDS promoted iron accumulation and ferroptosis through inducing ferritin autophagy in PTCL.
a Venn diagram showed the overlap between up- and down-regulated autophagy-associated molecules in RNA-seq and TMT-mass spectrometry. There were 61 (low, n = 24; high, n = 37) molecules with consistent expression change trend in RNA-seq and TMT-mass spectrometry. b, c The expression level of ATG2A and HSPB1 was regulated by AT56 treatment, PTGDS knockdown and overexpression at 36 h. d AT56 treatment for 36 h increased the expression of P62, Beclin 1 and LC3B in PTCL cells. e Chloroquine treatment for 48 h partly reversed the inhibitory effects of AT56 on the proliferation of PTCL cells. f, g The promoting role of AT56 on lipid ROS accumulation was partly reversed by chloroquine treatment for 36 h. h Chloroquine enhanced the role of AT56 on the expression of autophagy- and iron metabolism-associated proteins at 36 h. i The accumulation of Fe2+ caused by AT56 was reversed by chloroquine treatment for 36 h in PTCL cells. j Western blotting results showed the expression of autophagy-associated proteins in mouse model. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Firstly, to elucidate the regulatory of targeting PTGDS on autophagy process, we found that both AT56 treatment and PTGDS knockdown could increase the mRNA level of ATG2A, which was recognized to promote the formation of autophagosomes, while PTGDS overexpression decreased it (Fig. 7b). Meanwhile, the mRNA expression of HSPB1, a negative regulator of autophagy, was decreased by AT56 treatment and PTGDS knockdown, and increased by PTGDS overexpression in PTCL cells (Fig. 7c). Moreover, AT56 treatment and PTGDS knockdown increased the expression of key autophagy regulators, including P62, Beclin 1 and LC3B in PTCL cells (Fig. 7d, Supplementary Fig. S7A), indicating the increased formation of autophagosome and the activation of autophagy process.
Next, we performed experiments to decipher whether targeting PTGDS promoted ferroptosis through regulating autophagy in PTCL cells. Chloroquine, a classic autophagy inhibitor through suppressing the degradation in lysosomes, could partly reverse the inhibitory effects of AT56 treatment and PTGDS knockdown on the proliferation of PTCL cells (Fig. 7e, Supplementary Fig. S7B). Besides, the promoting role of AT56 on lipid ROS accumulation was partly reversed by chloroquine (Fig. 7f, g). Chloroquine treatment could also enhance the promoting role of AT56 treatment in the expression of autophagy-associated proteins (P62, Beclin 1 and LC3B) and iron metabolism-associated proteins (FTH1 and FTL) (Fig. 7h). Furthermore, the accumulation of Fe2+ caused by AT56 treatment and PTGDS knockdown was reversed by chloroquine in PTCL cells (Fig. 7i, Supplementary Fig. S7C). Besides, the regulatory role of targeting PTGDS on the expression of autophagy-associated proteins was confirmed in tumor tissue from mouse models (Fig. 7j). Overall, all the above results indicated that targeting PTGDS promoted iron accumulation and ferroptosis through inducing ferritin autophagy in PTCL
PTGDS interacted with HMOX1 to regulate iron metabolism and ferroptosis process in PTCLMolecular docking and interaction relationship analysis of PTGDS and differentially expressed ferroptosis-associated molecules in TMT-mass spectrometry and RNA-seq was performed to elucidate the precise mechanism through which targeting PTGDS promotes ferroptosis process in PTCL (Fig. 8a, Supplementary Fig. S8A). Among them, HMOX1 protein, the rate-limiting enzyme in the degradation of heme to release free iron, was found to possess potential interactions with PTGDS protein (Fig. 8a), and HMOX1 has been demonstrated to play key role in the process of ferroptosis and autophagy [22, 23]. Furthermore, results of confocal immunofluorescent and Co-IP verified the colocalization and interaction between PTGDS and HMOX1 in PTCL cells (Fig. 8b, c). Both AT56 treatment and PTGDS knockdown could increase the expression level of HMOX1 in PTCL cells (Fig. 8d, Supplementary Fig. S8B).
Fig. 8: PTGDS interacted with HMOX1 to regulate iron metabolism and ferroptosis process in PTCL.
a Molecular docking pattern diagrams of PTGDS protein (blue) and HMOX1 protein (red). Confidence score = 0.8. b, c The colocalization and interaction between PTGDS and HMOX1 were verified in PTCL cells through confocal immunofluorescent and Co-IP. Bar = 10 μm. d AT56 treatment for 36 h increased the expression level of HMOX1 in PTCL cells. e Western blotting results verified the transfection efficiency. f HMOX1 knockdown reversed the inhibitory role of AT56 on cell proliferation at 48 h. g, h The regulatory role of AT56 on the expression of ferroptosis- and autophagy-associated proteins, and Fe2+ accumulation was reversed by HMOX1 knockdown at 36 h. i Co-IP assay showed the interaction between PTGDS protein and HMOX1 protein with wild type and H25A mutation. j H25A mutation reversed the regulatory role of HMOX1 on the expression of ferroptosis- and autophagy-associated proteins. k Mechanism diagram summarized that PTGDS promotes the development of PTCL through regulating HMOX1-mediated iron metabolism and ferroptosis process, and targeting PTGDS could exert synergistic anti-tumor effects with ferroptosis inducers Erastin and Sorafenib in PTCL. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
To decipher whether HMOX1 was involved in the regulatory role of PTGDS on ferroptosis in PTCL, we performed lentiviral transfection to knockdown the expression of HMOX1 in PTCL cells, and the transfection efficiency was validated by western blotting (Fig. 8e). HMOX1 knockdown could reverse the inhibitory role of AT56 treatment on the proliferation of PTCL cells (Fig. 8f). Moreover, the regulatory role of AT56 on the expression of ferroptosis-associated and autophagy-associated proteins, and the accumulation of Fe2+ was reversed by HMOX1 knockdown in PTCL cells (Fig. 8g, h), indicating that HMOX1 played a key role in the positive effect of targeting PTGDS on iron metabolism and ferroptosis process in PTCL.
As cytoplasmic protein HMOX1 is mainly anchored on endoplasmic reticulum, the lack of C-terminal transmembrane segment could induce the translocation of HMOX1 protein to nucleus, which enhances the growth and invasiveness of tumor cells [24,25,26]. Our results showed that the treatment with AT56 had no effect on the cytosolic and nuclear distribution of HMOX1 protein in PTCL cells (Supplementary Fig. S8C). Furthermore, previous research revealed that HMOX1 with H25A mutation lost the catalytic activity to degrade heme and release free iron [27]. In our study, H25A mutation had no effect on the interaction between HMOX1 proteins and PTGDS proteins (Fig. 8i). Besides, wild type HMOX1 could regulate the expression of ferroptosis-associated and autophagy-associated molecules in PTCL cells (Fig. 8j), suggesting the promoting role of HMOX1 in the ferroptosis and autophagy process in PTCL. However, HMOX1 with H25A mutation displayed no such effect (Fig. 8j), indicating that targeting PTGDS promoted the process of ferroptosis and autophagy through enhancing the catalytic activity of HMOX1. Collectively, these results revealed that the interaction of PTGDS and HMOX1 could promote the progression of PTCL through inhibiting ferroptosis process, which was mediated by HMOX1-mediated heme degradation and ferritin autophagy (Fig. 8k).
留言 (0)