
記住我
Non-classical MHC class I genes encode for a heterogeneous group of molecules that in mammals have been shown to possess diverse functional roles (reviewed in (1)). Unlike classical MHC class I molecules which are expressed on the surface of most cells, many non-classical MHC class I genes have more restricted expression patterns (2, 3). Common to many non-classical MHC class I genes is the lack of the canonical S-X-Y promoter module which, when cooperatively bound by a multiplex protein complex, consisting of several transcription factors bound to NLRC5, drive transactivation of classical MHC class I genes in mammals (4, 5). Thus, differing from classical MHC class I genes, distinct transcriptional and post-transcriptional control mechanisms are involved in regulating basal and inducible expression of many non-classical MHC class I genes (6–11).
In salmonids, non-classical MHC class I L lineage genes are encoded outside the classical MHC class I core region and are found scattered across multiple chromosomes (12). Although salmonid L lineage genes share structural similarities with their classical MHC class I counterparts, the overall sequence identity is low and common to all L lineage sequences is a lack of canonical peptide anchoring residues and a relatively high hydrophobicity within the antigen binding groove (13, 14). In Atlantic salmon, low to modest constitutive expression of L lineage genes is evident in primary and secondary lymphoid tissues as well as in mucosal organs with some genes, such as Sasa-LFA, displaying a highly tissue restricted expression pattern (15). However, while basal L lineage mRNA expression is markedly lower, ranging between 50 to 100-fold lower compared to that of classical MHC class I (14, 15), during pathological conditions, such as viral and to a lesser extent bacterial infections, discrete L lineage gene transcription is induced (15). For example, intraperitoneal infection of Atlantic salmon parr with the salmonid alpha virus 3 (SAV3), an enveloped, single-stranded, positive-sense RNA virus, elicits induction of distinct L lineage genes implying that, Sasa-LIA but also, Sasa-LGA1 and to a lesser extent Sasa-LHA are involved in the early anti-viral immune response of Atlantic salmon (15). In support of this, type I IFNa1 induces Sasa-LIA and Sasa-LGA1 mRNA levels in Atlantic salmon kidney (ASK) cells (15). Moreover, IFNγ stimulation moderately modifies Sasa-LIA and Sasa-LGA1 gene expression in Atlantic salmon head kidney (SHK 1) cells (16) hinting at regulatory roles of both type I and type II IFNs in L lineage gene induction.
In mammals, it is well documented that type I IFNs induce expression of classical MHC class I genes and that a major component of the antiviral properties of type I IFNs is related to the interferon induced regulation of genes involved in processing and presentation of antigens (17–19). Type I interferon induced MHC class I transcription is mediated through both canonical and non-canonical JAK–STAT signaling pathways involving STAT1-STAT2 heterodimers and STAT1 homodimers and is dependent on IFN regulatory factors (IRFs), that bind to interferon stimulated response elements (ISRE) within the MHC class I promoters (20, 21). Similarly, IFNγ enhance expression of MHC class I through induction of IRF1, which in turn bind to ISRE elements in the MHC class I promoter (18). In fish, like in mammals, classical MHC class I is ubiquitously expressed and transcription is upregulated in response to viral infections (22, 23) and interferon stimulations (16). The promoter region of classical MHC class I in rainbow trout and Atlantic salmon, like that of higher vertebrates, contains an S-X-Y-like motif as well as a conserved ISRE element and multiple putative Gamma interferon activation (GAS-) and IRF sites, supporting IFN type I and type II -inducible properties (22, 24). Comparably, the promoter region of Atlantic salmon non-classical L lineage genes contains subgroup specific regulatory elements, which are potentially targeted by IFN regulatory factors (IRFs) and signal transducer and activator of transcription (STAT) proteins but lack a S-X-Y motif (15). These results are indicative of a large functional diversity among salmonid L lineage genes raising basic questions about how the expressions of these genes are regulated.
To date, type I IFN and type II IFN genes have been cloned and sequenced in several fish species and both types of IFNs have been produced as recombinant proteins, which have been shown to possess distinct biological activities (25–27). Atlantic salmon possess a broad repertoire of type I IFN genes encompassing at least 6 different classes (IFNa-IFNf) that may be classified into two groups based on the presence of two or four cysteines, forming one or two disulphide bonds, respectively (28). Among these, the 2-cysteine containing IFNa and the 4-cysteine containing IFNb and IFNc all induce expression of antiviral proteins and various transcription factors, including IRFs, albeit with different potentials (26). Comparably, fish IFNγ has been shown to induce expression of classical MHC class II (29) and classical MHC class I as well as a large number of genes encoding for proteins related to antigen processing and presentation (16). Like type I IFNs, rIFNγ also stimulates gene expression of the antiviral proteins Mx, suggesting some cross talk among the IFN signaling pathways (30).
While fish and mammalian IFNs have evolved quite differently and are not orthologs they appear to mediate their cellular responses in a similar manner utilizing cell specific receptors and activation of signaling pathways, predominantly dependent on sequential phosphorylation of the tyrosine residues of the Janus tyrosine kinases/signal transducers and activators of transcription proteins (JAK/STAT) which modulates the expression of specific genes and enhance antiviral mechanisms (reviewed in (31, 32)). Several Atlantic salmon type I IFN receptors, which likely show selectivity in binding affinity to different IFNs, have been identified, allowing for a large potential functional variation among the type I IFNs (33).
In this study, we undertook a comprehensive analysis detailing viral and interferon induced transcriptional induction patterns of Atlantic salmon L lineage genes, focusing on Sasa-LIA and Sasa-LGA1. Expression kinetics of L lineage genes were assessed both in vitro and in vivo following viral challenges, demonstrating that while both Sasa-LIA and Sasa-LGA1 are upregulated in response to SAV3 but not infectious pancreatic necrosis virus (IPNV) infection, there is a clear difference in kinetics and temporal regulation between these two genes indicating a tight regulatory control and functional diversification. Furthermore, characterization of type I and type II interferon induced transcription of Sasa-LIA and Sasa-LGA1 revealed specific induction patterns that are regulated by subgroup specific, evolutionary conserved promoter elements.
2 Materials and methods2.1 Experimental animals and infection protocolsSamples from SAV3-infected Atlantic salmon were obtained from an in vivo SAV3 challenge trial described in detail elsewhere (34). Briefly, rSAV3 (referring to SAV3 virus recovered from cells transfected with a plasmid containing the entire SAV3 genome (prSAV3; originally cloned from the wild-type SAV3 isolate H20/03 (DQ149204, AY604236)) was used. The same prSAV3 plasmid was used as a backbone for the construction of SAV3 infectious strains with targeted mutations, two of which (SAV3-E2319A and rSAV3-CapNLS) were included in this study. SAV3-E2319A has a mutation in the predicted N-linked glycosylation motif in the E2 protein while rSAV3-CapNLS has a mutation in the subcellular localization signals of the capsid protein (35). Both attenuated SAV3 strains are infectious, transmittable to naïve cohabitant fish and induce innate anti-viral responses (34).
The in vivo experiments were conducted at the Aquaculture Research Station, Tromsø, Norway. The challenge trials were approved by the Norwegian Animal Research Authority (NFDA) according to the European Union Directive 2010/63/EU (permit numbers 16409 and 19014) and were performed in accordance with the recommendations of the current animal welfare regulations: FOR-1996-01-15-23 (Norway). Briefly, non-vaccinated Atlantic salmon, pre-smolts, strain NLA, were reared in a hatchery at the Aquaculture Research Station, Tromsø, Norway and confirmed free of the salmon pathogens ISAV, SAV, PRV and IPNV by RT-RT-qPCR. Fish were kept in running freshwater at 10°C, exposed to continuous light and fed with commercial dry feed and starved for 24 hours prior to handling and sampling. The fish were randomly selected for immunization, anesthetized by bath immersion in benzocaine chloride (0.5 g/10 L) for 2–5 min, labelled (tattoo) and intramuscularly (i.m.) injected. In this study, samples from control fish i.m. injected with PBS and fish i.m. infected with 0.2 ml cell culture medium containing 102 TCID50 virus [rSAV3, rSAV3-E2319A or rSAV3-CapNLS (34)] or with a vaccine based on inactivated SAV3 with water-in-oil adjuvant (36) (kindly supplied by PHARMAQ AS) were analyzed. Shedder fish injected with SAV3 H20/03/2 at week 7 [as described in (34)], were added after sampling at week 8. Samples from 6-fish/time point were collected through a 12-week sampling period. Organs (heart, spleen, head kidney and gill) from virus-challenged and control fish were aseptically collected at 2, 4, 6-, 8-, 10- and 12-weeks post infection and kept in RNA-later. The organs were used for gene expression analyses by RT-qPCR following RNA isolation and subsequent cDNA synthesis as described below.
2.2 Cell lines and virusSSP-9 cells derived from Atlantic salmon head kidney (HK) (37), were kindly provided by Dr. S. Perez-Prieto (CSIC, Madrid, Spain). Chinook salmon embryo (CHSE-214) cells were purchased from American Type Culture Collection. Cell lines were maintained as monolayers in Leibovitz’s medium with L-glutamine (L-15) (Life Technologies) supplemented with antibiotics (10 U/ml penicillin, 10 μg/ml streptomycin) and 8% fetal bovine serum (FBS) at 20°C. For all in vitro infection trials Salmonid alphavirus subtype 3 (SAV3) (PDV-H10-PA3, provided by Professor Øystein Evensen, Norwegian University of Life Sciences) was used. The SAV3 virus was propagated in CHH-1 cells in L-15+ with 5% FBS at 15°C. Virus titer was determined by the TCID50 method. Infectious pancreatic virus (IPNV) [strain N1, serotype Sp (38)] were propagation in CHSE-214 cells and titrated as described in (39).
2.3 In vitro SAV3 and IPNV infectionSSP9 and CHSE-214 cells were seeded in 6-well plates at a density of approx. 2,5 × 105 cells/ml and grown until 70% confluency. Culture media was removed, and cells were washed two times with 2 ml sterile PBS followed by addition of serum and antibiotic free medium containing SAV3 or IPNV (MOI = 0,1, MOI= 1 or MOI= 5). After allowing the virus to be absorbed for 3 h, the medium was replaced with L-15+ supplemented with 2% FBS and cells were incubated for 1, 3, 5, 7, 9, 10 and 12 days following SAV3 challenge and 6, 12, 24, 48 and 72 h following IPNV infection. Variable time points for RNA isolation were based on documented differences in SAV3 and IPNV replication kinetics in CHSE-214 cells wherein in for IPNV, viral specific RNA synthesis peaks 8-10 hours after infection (40) typically resulting in significant (>70%) CPE by 48 hours post infection rendering expression analysis at later time points challenging and largely uninformative (41, 42). In contrast, SAV3 infection of CHSE typically result in detectable viral transcripts around 2 dpi with minor CPE observed at 6 dpi (41, 42). Following respective incubation time points, RNA was isolated for cDNA synthesis and RT-qPCR (described in detail below).
2.4 ReagentsRecombinant Atlantic salmon IFNa1 (GenBank accession no. DQ354152.1), IFNc (GenBank accession no. JX524153) and IFNb (GeneBank accession no. JX524152) were produced by transfection of sub confluent HEK-293 cells with IFN expression plasmids as previously described (25). Rainbow trout (Oncorhynchus mykiss) IFNγ was produced in E. coli (43) and protein concentrations were measured with a QuickStart Bradford protein assay kit (Bio-Rad) with bovine serum albumin as a standard. JAK I inhibitor was obtained from Calbiochem (CAS 457081-03-7).
2.5 Primary Head kidney cell isolation and stimulationTotal leukocyte populations from Head-kidney (HK) were isolated on Percoll (GE Healthcare) gradients following previously established protocols (44). Briefly, HK were sampled aseptically and kept in ice-cold transport medium (L-15 medium with 10 U/ml penicillin, 10 μg/ml streptomycin, 2% fetal bovine serum, 20 U/ml heparin) until homogenization on 100 μm cells strainers (Falcon). The resulting cell suspensions were layered on 25/54% discontinuous Percoll gradients and centrifuged at 400 × g for 40 min at 4°C. Cells at the interface were collected and washed twice in L-15 medium. Cells were counted using an automatic cell counter (Countess II Automated cell counter, Thermo Fisher, cat. nr. AMQAF1000). A total of 3 x 106 cells/ml were seeded in 1 ml L-15+ media supplemented with 8% FBS in 24 well plates (Nunclon Delta Surface, Thermo Scientific), stimulated with 500U of rIFNa, rIFNb, rIFNc or 10ng/ml IFNγ and cultivated at 16°C for 6, 12, 24 or 48 hours. Cells cultured in L15+/8% FBS media alone and harvested at the corresponding time points were used as controls. For inhibitory studies HKLs were isolated and stimulated as described above and incubated for 12 hours in the absence or presence of 15 nmol/ml of JAK I inhibitor, cells with inhibitor alone were included as controls. Following respective incubation time points, RNA was isolated for cDNA synthesis and RT-qPCR (described in detail below).
2.6 SSP9 and CHSE stimulationsSSP-9 cells (7× 105 cells/well) and CHSE cells (7 x 105 cells/well) were seeded in 1 ml culture media in 6 well culture plates. Cells in triplicate wells were stimulated with 1000 U/ml rIFNa, 1000 U/ml rIFNc or 10 ng/ml rIFNγ and harvested at different time points. For inhibitory studies SSP9 cells were stimulated as described above and incubated for 12 hours in the absence or presence of 15 nmol or 150 nmol of JAK I inhibitor, cells with inhibitor alone were included as controls. Following respective incubation time points, RNA was isolated for cDNA synthesis and RT-qPCR (described in detail below).
2.7 Western blotSSP-9 cells grown in 6 well plates as described above were stimulated with 10 ng/ml rIFNγ in the presence or absence of JAK I inhibitor were harvested at 12 hps, washed and lysed in M-PER (Mammalian protein extraction buffer) supplemented with HALT Protease inhibitor (Thermo fisher) and subjected to SDS-PAGE using NuPAGE Novex Bis-Tris 4–12% gels (Life Technologies). Proteins were transferred to PVDF membrane and incubated in blocking agent (Intercept Blocking Buffer, Licor) in TBS-T overnight at 4°. The membranes were incubated with either a polyclonal rabbit antibody prepared against Atlantic salmon Mx1 diluted 1/3000 (v/v) followed by an incubation with goat anti-rabbit Ig (H+L) secondary antibody conjugated to Alexa fluor 680 diluted 1/10000 (v/v) for 1 h at room temperature or actin 1/1000 (v/v) followed by incubation with a goat-anti-mouse Ig (H+L) secondary antibody conjugated to Alexa fluor 680 diluted 1/10000 (v/v) for 1 h at room temperature. After washing the membrane was imaged using Odyssey CLX infrared imaging system.
2.8 MicroscopyTo study the temporal progression of IPNV and SAV3 infections (up to 72 hours and five days, respectively) along with control (uninfected CHSE-214), 2,25 × 105 CHSE-214 cells were seeded in 35mm confocal dishes and allowed to adhere overnight. Culture media was removed, and cells were washed two times with 2 ml sterile PBS followed by addition of serum- and antibiotic free medium containing SAV3 or IPNV (MOI= 1). After allowing the virus to be absorbed for 3 h, the medium was replaced with L-15+ supplemented with 2% FBS and cells were incubated for 1, 3, 5 and 7 days following SAV3 challenge and 6, 12, 24 and 48 h following IPNV infection. Following respective incubation time points, cells were fixed. For fixation, PFA was used for twenty minutes followed by three washes with PBS. The cell membranes were stained with CellMask™ orange followed by NucSpot® Live direct dye for nuclear staining and stored until imaging. Images were acquired using a DeltaVision OMX V4 Blaze imaging system from GE Healthcare Life Sciences equipped with a 60X 1.42NA oil-immersion objective from Olympus; three sCMOS cameras; and lasers for excitation at wavelengths of 488 nm, 568 nm, and 642 nm. The exposure time and illumination power were adjusted to obtain a maximum of 10000 grayscale counts on the camera chip. A total of ten z-plane frames with z-steps of 100nm were collected on each imaging channel along the optical axis of the objective. The frames were automatically stitched into an 8×8 tile mosaic image by the built-in software package SoftWoRx.
2.9 Quantitative PCR and transcript analysisTotal RNA from tissues, primary leucocytes, SSP9 and CHSE cell lines were isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer`s recommendation and RNA was quantified using NanoDrop (ND 1000 Spectrophotometer). For RNA isolated from tissues and primary leucocytes 1000 and 500 ng respectively was treated using DNase I to remove all residual genomic DNA. Twenty microliter cDNA reactions were synthesized using TacMan reverse transcription reagents (Applied Biosystems) using random hexamer primers under the following conditions: 25°C for 10 min, 37°C for 30 min and 95°C for 5 min. cDNA samples were diluted 1:2 and stored at -20°C until use. For SSP9 and CHSE cells cDNA was synthesized using the QuantiTect RT kit (Qiagen) according to the manufacturer`s instructions with 500ng of RNA per 20 µl of reaction. cDNA was diluted 1:2 and stored at -20°C until use. Quantitative PCR (RT-qPCR) was run as 10ul duplicate reactions on a 7500 Fast Real-Time PCR systems (Applied Biosystems) according to standard protocol. All primers were validated prior to use and primer sequences are available upon request. For each primer pair and tissue/cell a negative control (no template) and a no reverse transcriptase control RT (-) was performed. A threshold difference of at least 6 quantification cycles (Cq) between Rt (+) and RT (-) was used as a cut-off. Ct values >38 was rejected. Parameters were as follows: 2 min 95°C followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 min. Melt curve analysis were performed to ensure that there were no artifacts, and a single product was amplified. Relative quantitative PCR gene expression analysis was performed using the ΔΔCt method. Expression of individual genes was examined relative to the endogenous EF1-α controlled. Relative expression (zero-hour samples) was calculated using the 2-ΔCt method. For infected tissues and stimulated cells, fold change or alternatively log2 fold change was calculated against the appropriate controls.
2.10 Plasmid construct designSasa-LGA1 and Sasa-LIA promoter regions containing 2000 bp upstream and 75 bp downstream of the transcriptional start site were extracted from the Salmo salar genome NC_027320, GCF_000233375.1. First, the transcription factor biding sites were identified using the online application PROMO which uses TRANSFAC database v8.3.0 and constructs positional weight matrices in a species or taxon using known transcription factor binding sites and then search for matches in query DNA sequences (45). Based on these results. The 2000 bp sequence was further processed to create different constructs for luciferase assay containing different promoters. All constructs were synthesized in PGL basic promoter by Twist Biosciences, San Francisco. The full 2000 bp upstream promoter was synthesized in PGL basic promoter for both Sasa-LGA and Sasa-LIA. For Sasa-LIA truncated promoter construct were designed to either including or exclude specific ISRE and GAS elements; -150/+75 including the ISRE and GAS elements, along with -700/+75 bp, -1200/+75 bp, -1200/-700 bp (including +75 at 5’). The mutated constructs were focused on only ISRE and GAS elements. The Sasa-LIA-150 + 75 bp construct was selected to mutate ISRE element GAAAGTGAAA to CCGAGTGACG and GAS- like element TTCAGAA to GCGAGCG. Similarly, for Sasa-LGA a systematic deletion of promoter was done with +75-200 including the IRF1/3 and ISRE element, +75-500 including STAT binding site, -1200/+75, and -1300/-1900 (including +75 at TSS) that included putative GAS like element but no proximal ISRE elements. For mutation, the ISRE element in the -200/+75 construct was mutated.
2.11 Luciferase assayCHSE-214 were seeded into 96-well culture plates at a density of 1.6 × 104 cells/well in 100ul of L-15 medium supplemented with 8% FBS and grown to 50% confluence overnight at 20 °C. The cells were transiently transfected by adding a transfection mix consisting of 10 µl Opti-MEM® (Life Technologies) solution containing 90 ng of promoter reporter (firefly luciferase) construct, 10 ng Renilla luciferase vector (Promega- Madison WI), and 0.3 µl TransIT-LT1 per well. Atlantic salmon LIA and LGA1 promoter constructs were investigated, while pGL3-basic was included as empty vector control. All promoter fragments and mutants were synthesized by Twist bioscience and cloned into pGL3-basic. 24 hours post transfection, cells were stimulated with 600 U/ml of recombinant (r)Type I rIFNa or rIFNc or 30 ng/ml of rINFγ. Luciferase activity was measured 48 h post stimulation using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) according to the manufacturer’s protocol. The constitutively expressing Renilla luciferase construct provided an internal control value to which the expression of the experimental firefly luciferase was normalized. The firefly and Renilla luciferase activity was measured in a Luminoscan RT luminometer, in which all samples for the luciferase assay were set up in four parallels for each treatment and the results are given as relative light units (RLU). The results are presented as fold change in relative light units (RLU) by dividing the RLU of the stimulated samples by the average RLU of the corresponding non-stimulated samples.
2.12 Statistical analysisFor in vivo studies all quantitative data were based on duplicated measurements from a minimum of four fish n ϵ (46) and were analyzed in GraphPad Prism 8. For primary cells and cell lines all quantitative data were based on triplicated samples undergoing duplicated measurements. All in vitro experiments were repeated a minimum of three times in independent experiments. Statistical evaluations were performed using Tukey`s multiple comparisons test following a significant one-way ANOVA. Correlation among MHC class I L lineage expression, pathogen load and interferon expression were determined using the Pearson Correlation coefficiency (p=0.05) calculated from the relative expression of each gene normalized to EF1-alpha(B). For all analysis a p value < 0.05 was considered significant.
2.13 Data mining and phylogenetic analysisGenome searches were performed using previously identified Atlantic salmon MHC class I L lineage LIA and LGA1 sequences and blasted against annotated salmonid genomes available in NCBI using mega blast. Genomic regions identified through these searches were screened for LIA and LGA1 leader sequences based on Atlantic salmon LIA and LGA1 cDNA sequences and the upstream proximal (-500) promoter regions were extracted. Genomes used in this study were as follows: Oncorhyncus gorbuscha GCA_021184085.1 (pink salmon), Oncorhyncus keta GCA_023373465.1 (chum salmon), Oncorhynchus kisutch GCA_002021735.2 (coho salmon), Oncorhynchus mykiss GCA_013265735.3 (rainbow trout), Oncorhynchus nerka GCA_006149115.2 (sockeye salmon), Oncorhynchus tshawytscha GCA_018296145.1 (Chinook salmon), Salmo salar GCA_905237065.2 (Atlantic salmon), Salmo trutta.
GCA_901001165.1 (brown trout), Salvelinus GCA_002910315.2 (unclassified species in the genus Salvelinus), Salvelinus fontinalis GCA_029448725.1 (brook trout) and Salvelinus namaycush GCA_016432855.1 (lake trout). For promoter regions, all evolutionary analyses were conducted in MEGA7. Sequences were aligned using ClustalX with manual corrections for some predicted sequences and bootstrapped phylogenetic trees were constructed using the Maximum Likelihood method. The percentage of trees in which the associated taxa clustered together are shown next to the branches. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site. Gene models were predicted based on expressed LIA and LGA1 sequences in Atlantic salmon.
3 Results3.1 Non-classical MHC class I L lineage gene expression profiles following in vivo SAV3 challengeTo further delineate gene specific SAV3 inducible responses among Atlantic salmon L lineage genes, tissue specific transcriptional induction and gene expression kinetics of six functionally expressed L lineage genes (Sasa-LIA, Sasa-LGA1, Sasa-LHA, Sasa-LDA, Sasa-LFA and Sasa-LCA) (15) were analyzed over a 12-week experimental immunization challenge using a recombinant SAV3 infectious strain. Accordingly, fish were i.m. infected with an infectious recombinant (r)SAV3 strain and subsequently challenged on week 8 by co-habitation with SAV3 (H20/03) infected shedder fish as previously described (34). At different times post-infection (2-, 4-, 6-, 8-, 10- and 12-weeks post infection [wpi]), log2 fold change in expression of the various L lineage genes in the heart (main site of viral replication), head-kidney, spleen and gill were determined (Figure 1; Supplementary Figure 1). Consistent with previous reports (15) Sasa-LIA and Sasa-LGA1 gene expressions were markedly upregulated at the fist, 2 wpi, time point. For both Sasa-LIA and Sasa-LGA1 the highest level of induction was observed in the heart, with an average fold increase of 46 (SD ± 10) and 25 (SD ± 3.3) respectively, followed by head-kidney and to a lesser extent spleen, with little to no transcriptional induction observed in the gill (Figures 1B, C). Notably, Sasa-LIA gene expression in the heart, despite the continuous detection of viral specific transcripts, was transient (Figures 1B, C). Across all examined tissues Sasa-LIA gene expression peaked at the 2-week time point followed by a return to near baseline levels by 4 wpi. In stark contrast, Sasa-LGA1 expression in the heart remained elevated through the course of infection, albeit with a gradual decrease from 2 to 8 wpi. Similarly, 2 weeks after exposure to SAV3 infected shedders a moderate but transient induction of Sasa-LIA was observed in the heart and head-kidney with expression returning to baseline levels by the next sampling point. Comparably, post challenge Sasa-LGA1 expression in the heart was significantly upregulated compared to PBS controls at both the 10- and 12-week time points. Neither Sasa-LHA nor Sasa-LDA were upregulated with a >2 log2-fold increase following SAV3 infection in any of the tissues examined with the exception of heart where significant upregulation of both genes was apparent at 4 wpi (Figure 1E, Supplementary Figure 1). For Sasa-LCA significant upregulation was observed in the heart at 4- and 6-wpi, which was reciprocated 4 weeks after addition of shedder fish (Figure 1E. Supplementary Figure 1). Consistent with previous results indicating a highly tissue specific expression pattern for some L lineage genes (15) Sasa-LFA expression was only detectable above threshold levels in the gills and showed no significant transcriptional response to SAV3 challenge. In the same sample set expression patterns of IFNa, IFNc and IFNγ were examined (Figure 1E, Supplementary Figure 1) revealing, as expected, a strong induction of type I IFNa, IFNc and type II IFNγ in response to SAV3 infection. Similar to what was observed for Sasa-LIA, IFNa expression in the heart peaked at 2 wpi, was significantly reduced by 4 wpi and continued to decline at the 6-, and 8 wpi time points. In contrast, elevated expression levels of both IFNc and IFNγ were maintained at the 2-, 4- and 6-wpi time points followed by a marginal reduction at 8 wpi.
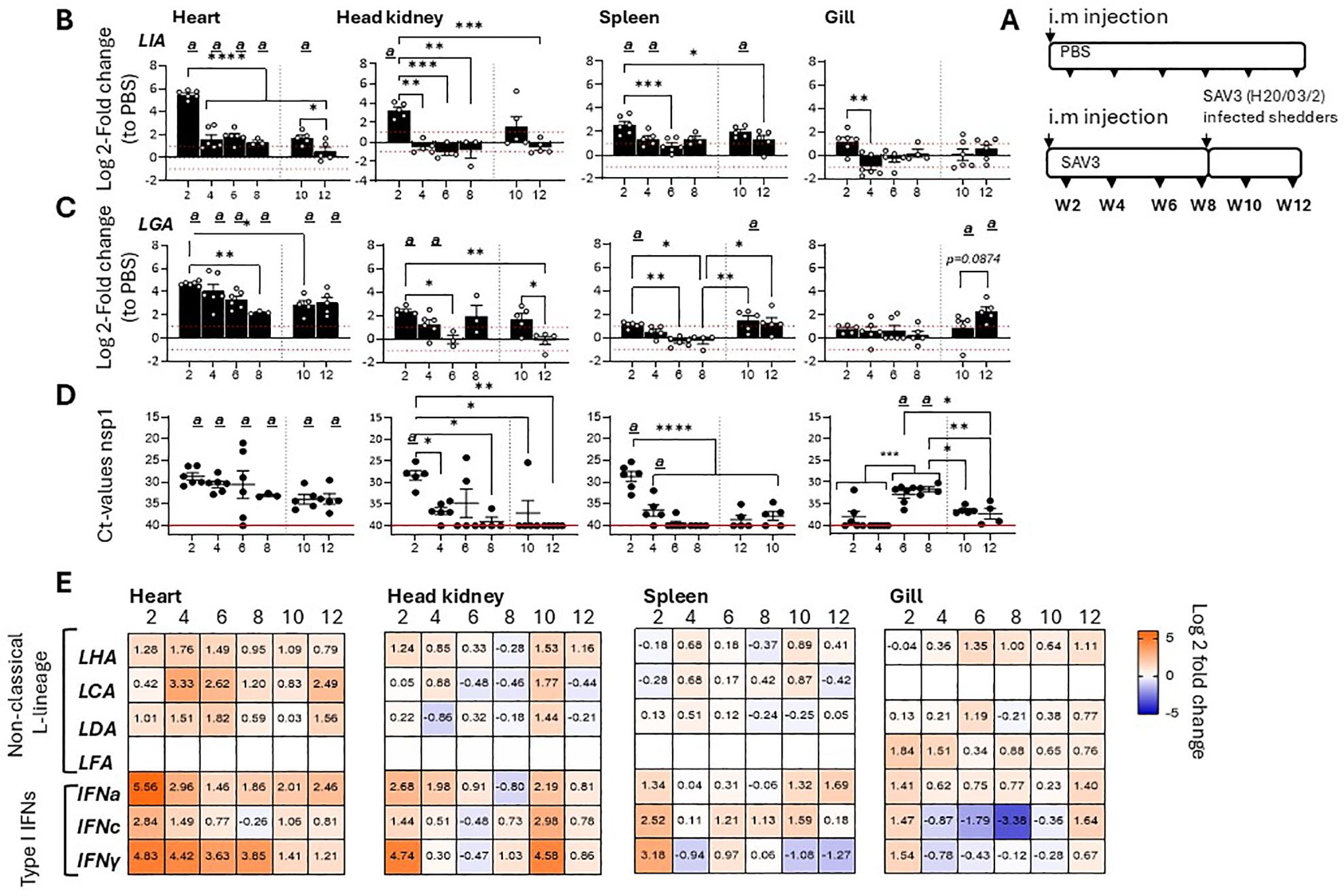
Figure 1. Sasa-LIA and Sasa-LGA1 are differentially regulated in response to SAV3 infection in vivo.(A) Schematic outline of the experimental setup indicating various sampling points and experimental groups. Log2 fold change in gene expression of L lineage MHC class I genes (B) Sasa-LIA and (C) Sasa-LGA1, normalized to the reference gene EF1α, in different tissues at various times post challenge (n ϵ 3-6), relative to the control groups from the same time point (n ϵ 6) is shown. Each dot represents an individual fish, the red dotted line intersecting the y-axis at 1 and -1 in (B) and (C) represent a 2-fold increase compared to PBS injected controls, significant (p < 0.05) upregulation compared to the corresponding control group is indicated by (a) and asterisks indicate the strength of significance among the different time points as indicated, *p < 0.05, **p < 0.01, ***p = 0.0001, ****p < 0.0001. (D) Individual Ct values and mean value of non-structural protein 1 (nsp1) detected by qPCR in heart, head-kidney spleen and gill at each sampling from 2- 12 wpi. (E) Heat maps illustrating average expression ratios of L lineage MHC class I genes (Sasa-LHA, Sasa-LCA, Sasa-LDA and Sasa-LFA), type I IFNa, type I IFNc and type II IFNγ as log2 fold-change values of the SAV3 infected groups relative to PBS injected controls for the indicated genes as measured by qPCR (bar graphs illustrating individual variation for each of the genes listed is shown in Supplementary Figure 1).
To investigate the factors governing discrete Sasa-LIA and Sasa-LGA1 transcriptional induction patterns we examined the expression of these genes in fish i.m. infected with two genetically modified infectious strains of rSAV3 or with a vaccine based on inactivated SAV3 with water-in-oil adjuvants (36) (Figure 2A). These rSAV3 strains, which have previously been shown to actively infect and replicate in Atlantic salmon (34) are attenuated in either the envelope protein E2 (rSAV3-E2319A) or the capsid protein nuclear localization signal (rSAV3-CapNLS). Similar to rSAV3, at 4 wpi both attenuated strains, as assessed by mRNA expression of select interferon stimulated genes, induce comparable anti-viral responses in the heart (34). Congruent with these observations, comparable levels of viral transcripts were detected in hearts isolated from fish infected with WT-rSAV3, rSAV3-E2319A or rSAV3-CapNLS while no viral transcripts were detected in fish injected with the inactivated SAV3 (Figure 2D). While inactivated SAV3 failed to induce Sasa-LIA gene expression, significantly increased mRNA levels were detected at 2 wpi in response to WT-rSAV3, rSAV3-E2319A and rSAV3-CapNLS (Figure 2B). However, Sasa-LIA expression in fish infected with rSAV3-E2319A was significantly lower compared to both rSAV3-CapNLS and WT- rSAV3. As seen in Figure 2C Sasa-LGA1 was upregulated in response to all three viral strains and, albeit not statistically significant, by 4 wpi inactivated SAV3 infection resulted in elevated Sasa-LGA1 expression comparable to that of fish infected with actively replicating SAV3 strains. As a comparison no significant difference in expression of type I IFNa, type II IFN-y or the interferon inducible gene Mx1/2 were observed between rSAV3, rSAV3-E2319A or rSAV3-CapNLS at 2 wpi (Figures 2E-G). However, at 4 wpi expression of IFNa in the rSAV3-E2319A was lower compared to the other viral strains and did not reach significant induction levels compared to fish injected with inactivated SAV3.
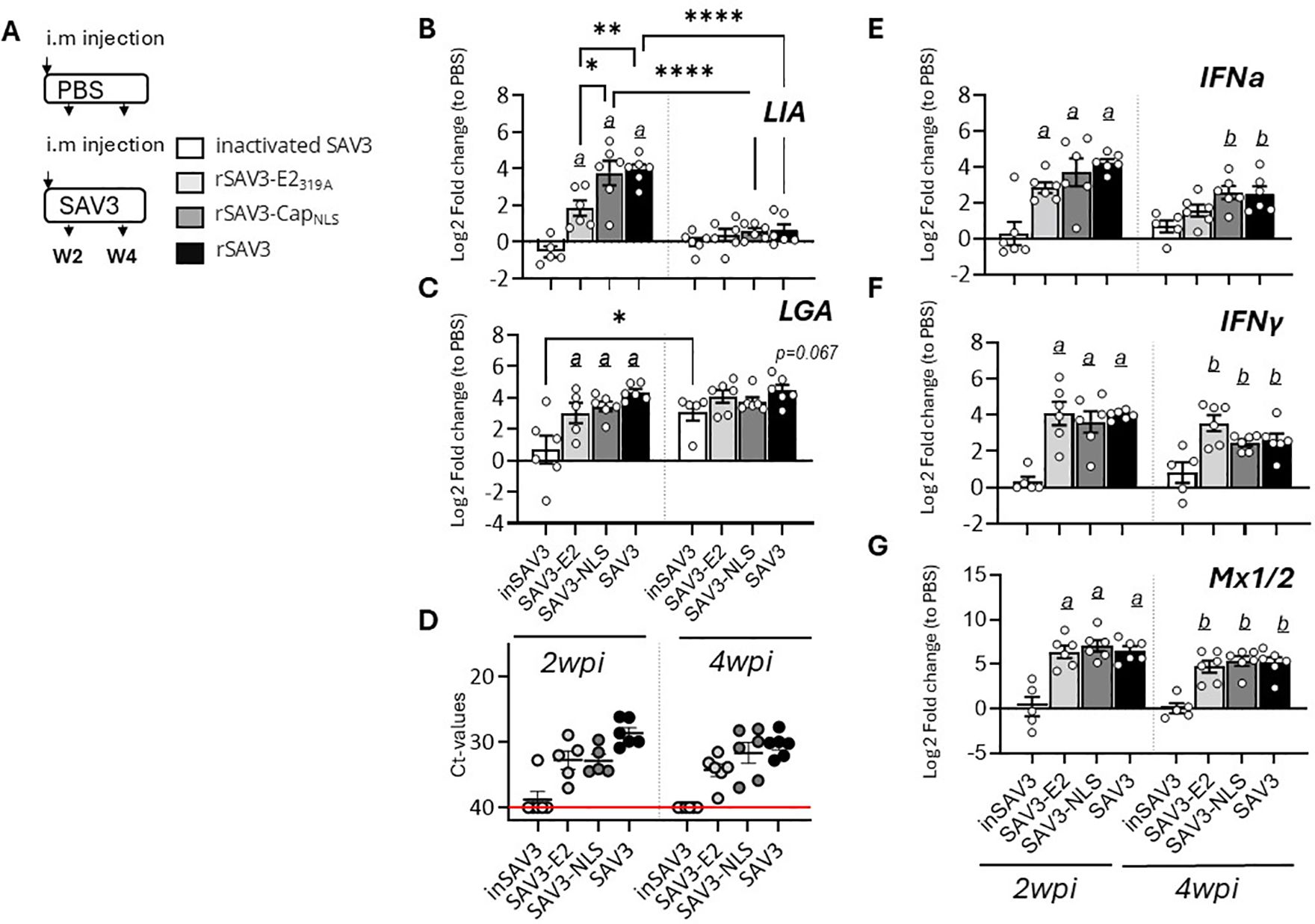
Figure 2. Sasa-LIA and Sasa-LGA1 expression in heart following infection with attenuated and inactivated SAV3. (A) Schematic outline of the experimental setup indicating various sampling points and experimental groups. (B-C, E-G) Log2 fold change in gene expression of Sasa-LIA, Sasa-LGA1, type I IFNa, type II IFNγ gamma and Mx1/2, normalized to the reference gene EF1α, at various times post challenge (n ϵ 6), relative to the control groups from the same time point (n ϵ 6) is shown. Asterisks indicate the strength of significance among the different time points as indicated, *p < 0.05, **p < 0.01, , ****p < 0.0001. Letters (a and b) above the bars in (B-C, E-G) indicate statistical significance compared to the group injected with inactivated SAV3 vaccine at 2- and 4-wpi respectively while (c) above the bars indicate statistical significance compared to the PBS injected group. (D) individual Ct value and mean value of nsp1 in in heart of groups infected with the different viral strains at 2- and 4wpi.
3.2 SAV3-infection upregulates LIA gene expression in vitro while IPNV infection has limited effectTo further assess the effects of viral infection on the regulation of L lineage gene expression, specifically LIA and LGA1, we analyzed the temporal dynamics of these genes in response to SAV3 infection in SSP-9 cells (37). Similar to in vivo observations Sasa-LIA gene expression was upregulated during the early stage of infection (1 dpi) with infection using a higher virus MOI resulting in higher Sasa-LIA induction (Figure 3A). Further, despite highest detection of SAV nsp1 RNA at 3 dpi, Sasa-LIA transcriptional levels returned to near baseline by this time point and remained at this level throughout the course of the infection (Figures 3A, C). A similar gene expression pattern was observed for IFNa which was strongly correlated with that of Sasa-LIA (Figure 3D, Supplementary Figure 2). In contrast to what was observed following in vivo SAV3 challenge Sasa-LGA1 was not significantly induced in response to SAV3 infection in SSP-9 cells (Figure 3B).
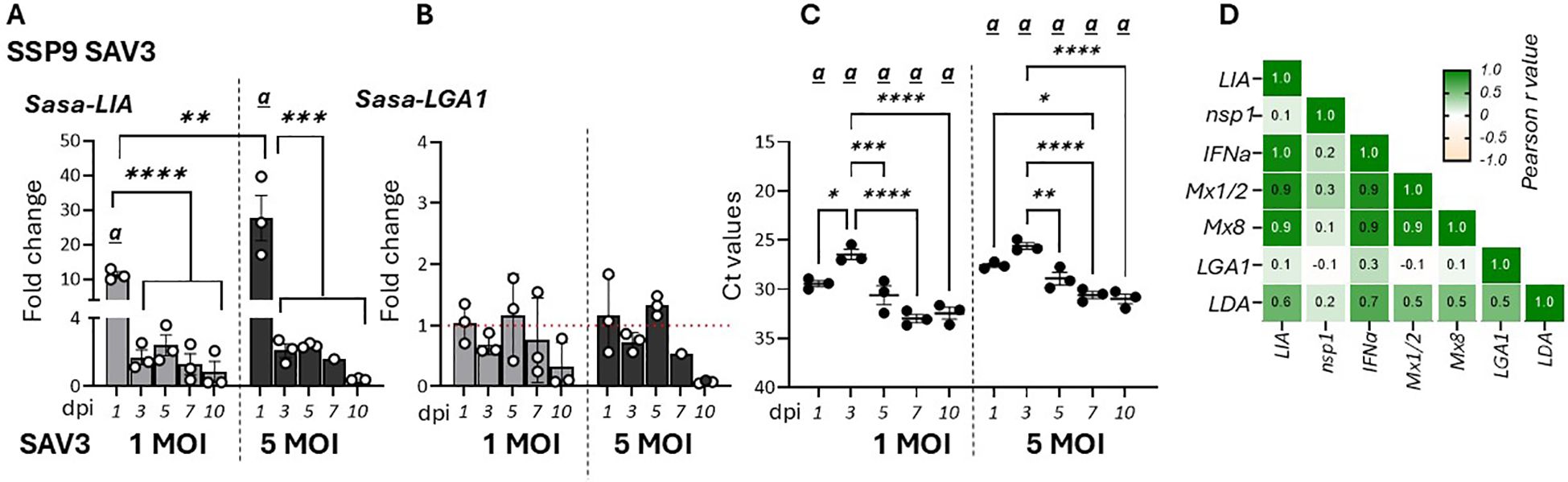
Figure 3. Sasa-LIA gene expression is transiently upregulated in SAV3 infected SSP9 cells. Relative gene expression of (A) Sasa-LIA and (B) Sasa-LGA1 measure 1-10 days post SAV3 in vitro infection in SSP9 cells (MOI = 1 or MOI = 5). Bars represent mean ± SE (n = 3/time point with individual results shown as white dots) expressed as fold induction compared to non-infected cells at each time point. All samples were analyzed with RT-qPCR and normalized against EF1α as reference gene. (C) Individual Ct values and mean values of nsp1 at the different time points following infection. Significant (p < 0.05) upregulation compared to the corresponding control group is indicated by (a) and significant differences among the different time points are indicated, *p < 0.05, **p < 0.01, ***p = 0.0001, ****p < 0.0001. (D) Correlation matrix showing mRNA expression correlation of select genes. Numbers in boxes indicate R values of analyzed gene pairs and the strength of correlation is colored as depicted in the scale bar. (Relative gene expression of ache genes in the correlation matrix can be found in Supplementary Figure 2).
Next, transcriptional responses of L lineage gene expression in response to viral infections in the chinook salmon embryonic cell line CHSE-214 were investigated. Here it should be noted that L lineage gene profiles in salmonids is complex, with a remarkably high degree of species-specific adaptations both in gene numbers and functionality (12). Thus, the L lineage profile of chinook salmon was contrasted with that of Atlantic salmon, revealing that while LIA is highly conserved and present as a bona-fide functionally expressed gene in both species, LGA1 has become pseudogenised in Chinook salmon (Figure 4A, Supplementary Figure 3). Consistent with the genomic data no expression of Onts-LGA1 were observed in CHSE-214 cells (data not shown). Accordingly, only Onts-LIA was examined further. Compared to SSP-9 cells, for CHSE-214 exposed to SAV3 at MOIs of 1 and 5 viral kinetics was different likely reflecting the genetic differences inherent to these two cell types. In CHSE-214 cells, Onts-LIA transcript levels were not significantly induced until around 7 dpi reaching a peak of induction at 9 dpi before declining by 12 dpi (Figure 4C). However, similar to what was observed in SSP-9 cells Onts-LIA mRNA levels were highly correlated with nsP1 and IFNa transcript levels, as well as with all representative interferon stimulated genes (ISGs) tested (Mx1/2, Mx8 and CXCL10) while no significant correlation was manifested between Onts-LIA and IFNc transcripts (Figure 4J, Supplementary Figure 4). When contrasted with the presence of SAV3 virus, as inferred from detection of nsp1 transcript levels, measured by RT-qPCR, viral transcripts could be detected already at day 3, reaching a peak at 7-9 dpi before declining at the 12 dpi time point (Figure 4E). Similarly, a gradual increase in CPE monitored via confocal microscopy on fixed cells, using cell mask and nuclear staining was detected for cells infected with 1 MOI SAV3 with signs of infection apparent as early as 1 dpi. Throughout the infection, compared to control cells, infected cells displayed distinct morphological changes including compromised cytoskeleton, cell membrane permeabilization, rounding and loss of cell adhesion (Figures 4F-I). In conclusion, these data indicate that while SAV3 infection leads to upregulation of Onts-LIA gene expression, transcriptional regulation appears to be tightly linked, possibly directly mediated by interferon stimulation rather than direct viral recognition.
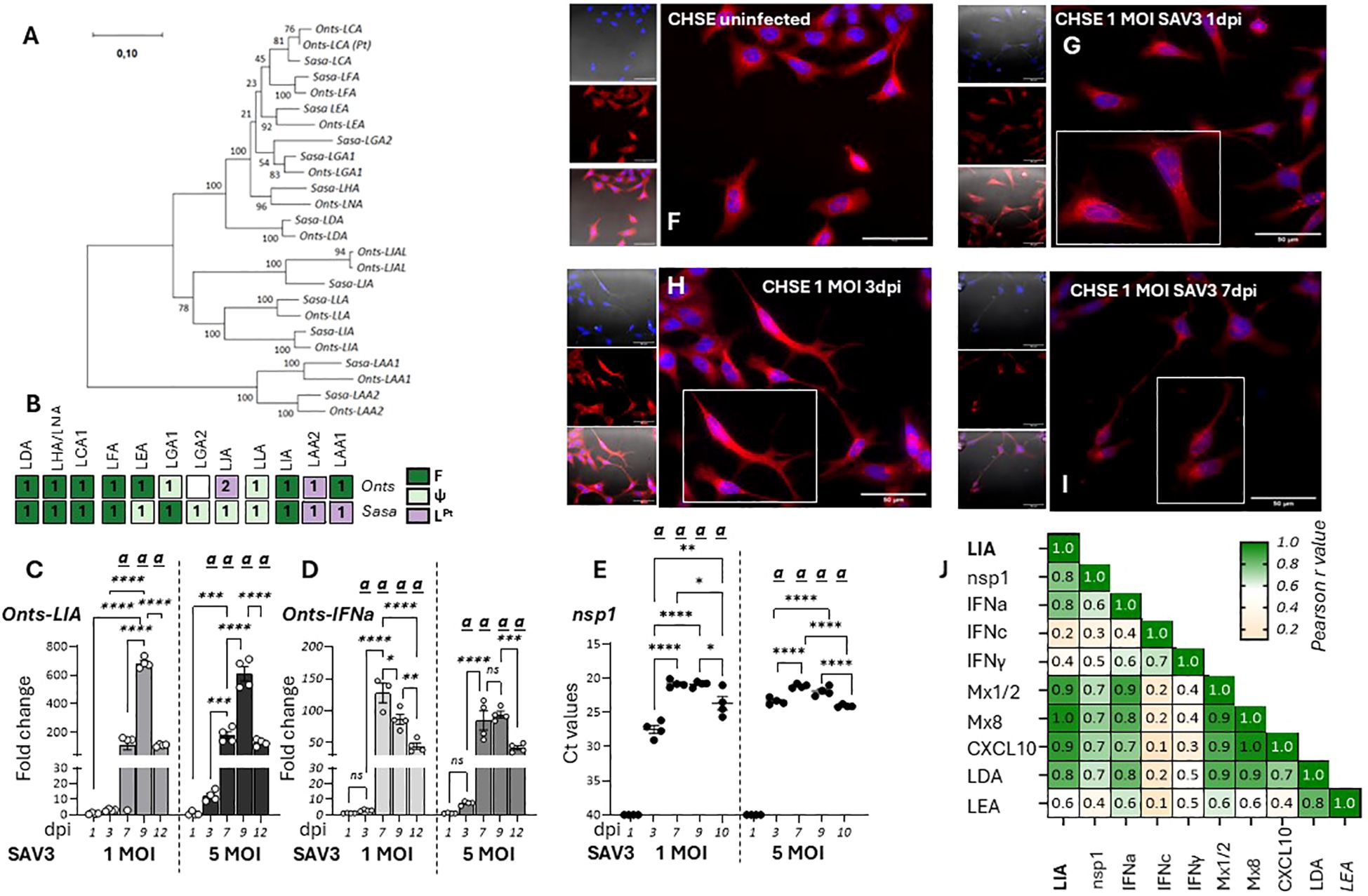
Figure 4. Onts-LIA is highly expressed in SAV3 infected CHSE cells. (A) Phylogenetic tree illustrating the relationships among Atlantic salmon and Chinook salmon L lineage genes. The tree with the highest likelihood is shown, and is drawn to scale, with branch lengths representing the number of substitutions per site. (B) Schematic representation summarizing the distribution and number of full length bona fide functional (F, dark green), pseudo (ψ, light green) and partial (LPt,purple) L linegae genes in the two species. Relative gene expression of (C) Onts-LIA and (D) Onts-IFNa measure 1-12 days post SAV3 in vitro infection in CHSE cells (MOI = 1 or MOI = 5). Bars represent mean ± SE (n = 4/time point with individual results shown as white dots) expressed as fold induction compared to non-infected cells at each time point. All samples were analyzed with RT-qPCR and normalized against EF1α as reference gene. SAV3 progression was estimated by (E) individual Ct values and mean values of nsp1 at the different time points following infection and (F-J) deconvolution microscopy. Representative images of control (F) and 1 MOI SAV3 infected CHSE-WT at (G) 1 dpi (H). 3dpi and (I) 7 dpi. Cells were stained with membrane stain CellMask™ Orange (red) and cytosolic stain NucSpot® Live 488 (Nunc488) (blue). The scale bar indicates 50μm. Morphological distinctions (CPE) were detected in CHSE cells at 1, 3, and 5dpi. (J) Correlation matrix showing correlation of mRNA expression of select genes throughout the experiment. Numbers in boxes indicate R values of analyzed gene pairs and the strength of correlation is colored as depicted in the scale bar. Significant (p < 0.05) upregulation compared to the corresponding control group is indicated by (a) and significant differences among the different time points in (C-E) are indicated, *p < 0.05, **p < 0.01, ***p = 0.0001, ****p < 0.0001. The infection experiment was performed twice with reproducible results.
In contrast to SAV3 which has been shown to induce the expression of type I IFN and ISGs both in vitro and in vivo IPNV, a dsRNA virus, inhibits IFN-induced responses in CHSE-214 cells (46). An interesting question was therefore how infections with IPNV would potentially influence the expression of L lineage genes. For CHSE-214 cells exposed to 1 MOI IPNV, CPE monitored via confocal microscopy showed a progressive loss of cell adhesion and compromised cell membrane and cytoskeleton along with increasing levels of lysed cell debris (Figures 5B–E). Signs of cell stress was apparent already at 6 hpi and progressively increased throughout the course of the infection. In concordance with the microscopic analysis Figure 5A shows IPNV VP2 transcript levels in the infected cells, which increased over time, indicating a productive infection. However, in stark contrast to SAV3 infections of the same cells, no significant induction of Onts-LIA was observed as compared to control cells. The mRNA expression of IFNa and Mx paralleled Onts-LIA expression and in general, expression of all three genes were very low throughout the study.
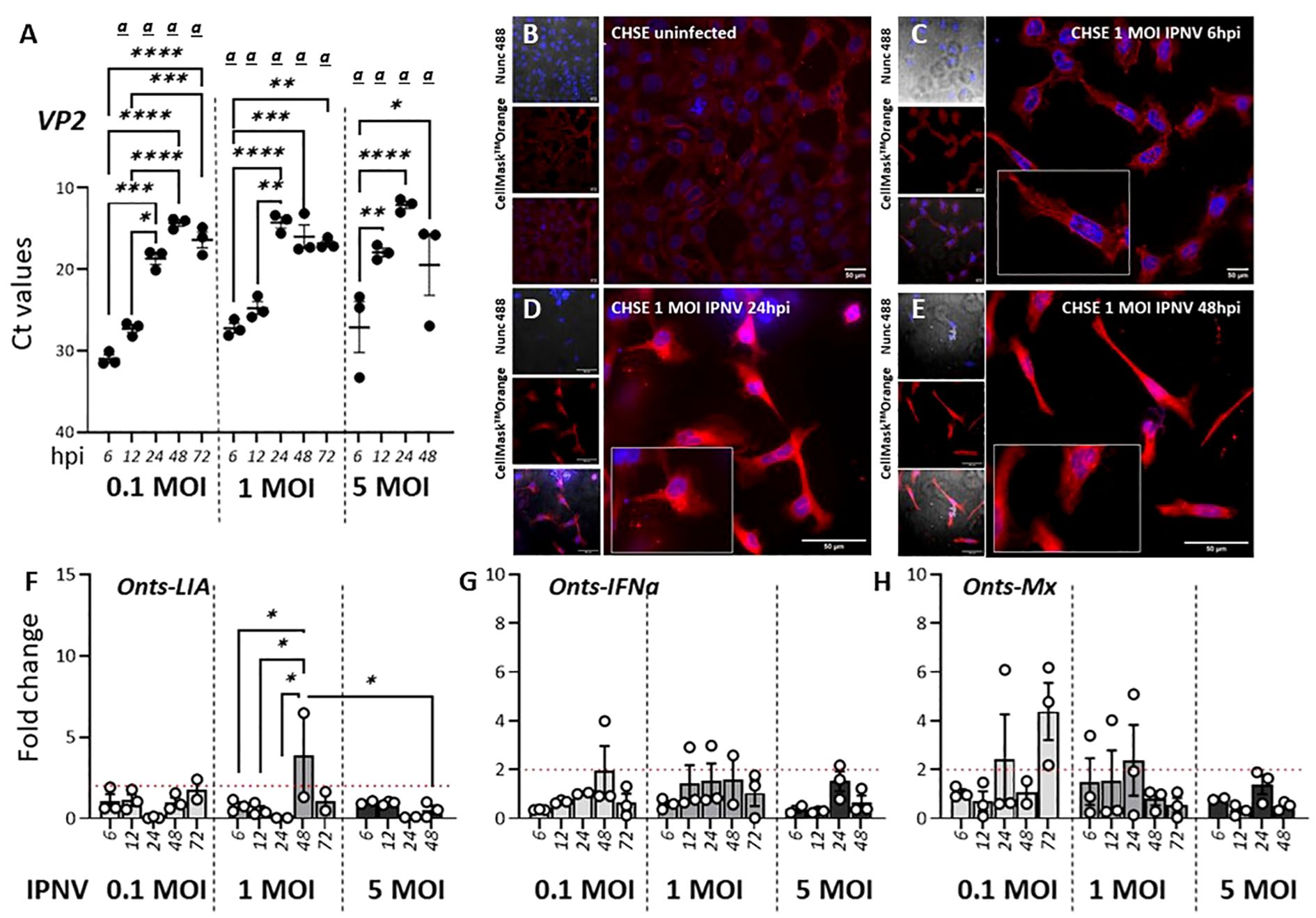
Figure 5. IPNV infection has a negligible effect on Onts-LIA expression. (A–D) Representative images of uninfected (A) and 1 MOI infected CHSE-214 cells at (B) 6 hours post infection (hpi) (C) 24 hpi and (D) 48hpi. Cells were stained with membrane stain CellMask™ Orange (red) and cytosolic stain NucSpot® Live 488 (Nunc488) (blue). The scale bar indicates 50μm. Morphological distinctions (CPE) were detected in CHSE cells at 6, 24 and 48hpi, upscaled in inserts. (E) Detection of viral VP2 mRNA by RT-qPCR following infection with 0,1, 1 or 5 MOI IPNV. RNA extracted from cells at MOI 5 at 72 h was, because of extensive CPE, of poor quality and no data from this time point is therefore represented in the figure. The relative gene expression of (F) Onts-LIA, (G) Onts-IFNa1 and (H) Onts-Mx were measured 6–72 h post in vitro infection in CHSE-214 cells with IPNV (MOI = 0.1, 1 and 5). Bars represent mean ± SE (n = 3/time point with individual results shown as white dots) expressed as fold induction compared to non-infected cells. Significant (p < 0.05) upregulation compared to the corresponding control group is indicated by (a) and significant differences among the different time points in are indicated, *p < 0.05, **p < 0.01, ***p = 0.0001, ****p < 0.0001.
3.3 Sasa/Onts-LIA and Sasa-LGA1 expression is differentially induced by representative type I and type II IFNsGiven that type I IFN classes are known to differ in their responses, we compared the ability of recombinant representatives of group 1 (rIFNa1) and group II (rIFNb and rIFNc) type I IFNs to modulate L lineage gene expression in primary head kidney leucocytes (HKLs, Figure 6), SSP-9 (Figures 7A, B) and CHSE-214 cells (Figure 7C). Further, to obtain additional insight into the interplay among type I and type II interferon induction and transcription regulation of L lineage genes, the modulating abilities of representative type I IFNs was contrasted with that of recombinant type II IFN (rIFNγ). For HKLs, expression of the interferon stimulated genes Mx1/2 and Mx8, which have been shown to differentially respond to IFNa and IFNγ stimulation (30) were included as controls and the transcriptional induction of IL-1, TNFα, IFNa1 and IFNγ was investigated in parallel (Supplementary Figure). In HKLs, 12 hours post stimulation, both Sasa-LIA and Sasa-LGA1 were strongly (~16 fold) upregulated in response to rINFγ stimulation (Figures 6A, B). Sasa-LIA expression was also significantly upregulated in response to type I rIFNa1 and rIFNc stimulation, while Sasa-LGA1 showed a modest induction in response to rIFNa1 and no significant induction in response to rIFNc stimulation at this time point. Comparably, while rIFNb stimulation induced expression of Mx1/2 and Mx8 at comparable levels to that of rIFNa1, no induction of L lineage gene expression was observed in response to rIFNb stimulation (Figure 6, Supplementary Figure 5). A modest induction of Sasa-LHA gene expression was observed in response to rINFγ with no significant induction in response to any of the type I IFNs tested. Sasa-LDA did not respond to type I IFNs or type II IFN stimulation and neither Sasa-LCA nor Sasa-LFA transcripts were detected in HKLs above threshold levels. Comparing the induction kinetics of Sasa-LIA and Sasa-LGA1 genes revealed that both L lineage genes responded rapidly, albeit with different magnitudes and distinct kinetics following stimulation with either rIFNa1, rIFNc or rIFNγ (Supplementary Figure 5). Significantly elevated transcript levels were detected as early as 6 hps, peaking between 12 and 24 hps, followed by a return to baseline by 48hps. Induction kinetics were different depending on the stimuli, with rIFNa1 resulting in a somewhat bimodular upregulation peaking at 6 and 24hps. Comparably, highest transcription levels in response to INFc was detected at 6 hps for both Sasa-LIA and Sasa-LGA1 while IFNγ stimulation resulted in continuous increasing levels of Sasa-LIA up on till the 24h time point while Sasa-LGA1 transcription levels peaked at the 6-hour time point.
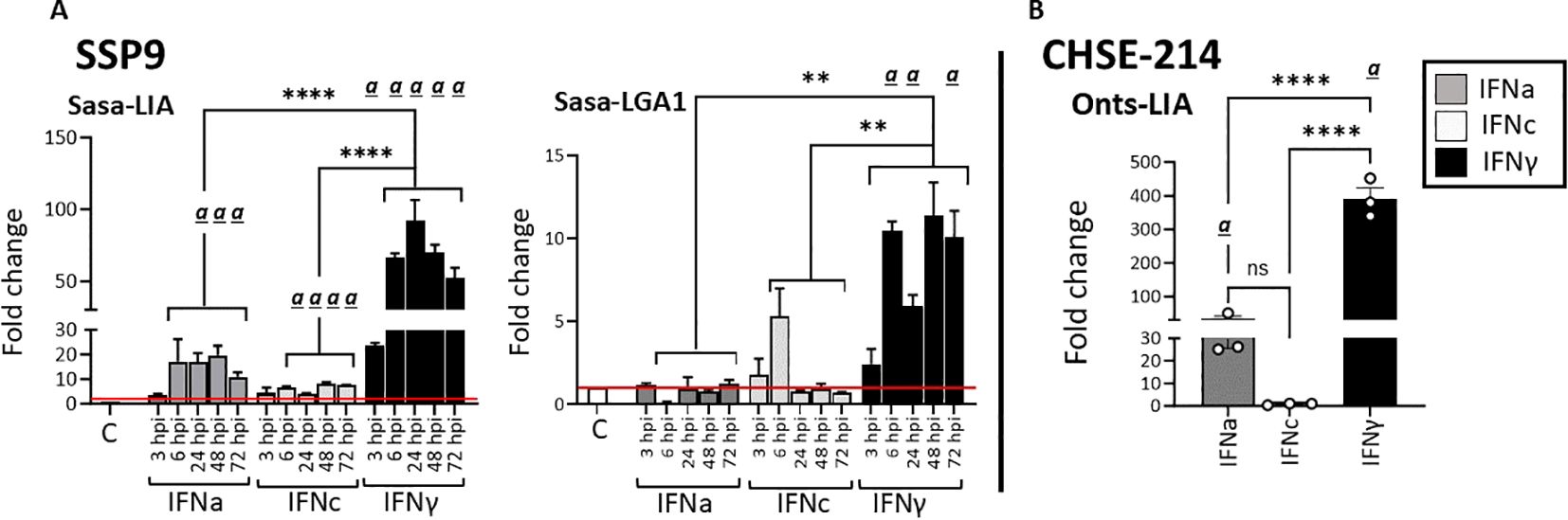
Figure 6. Type I (IFNa, IFNb and IFNc) and type II interferon stimulation results in variable induction of Sasa-LIA and Sasa-LGA in HKLs. Expression of MHC class I L lineage (A) Sasa-LIA and (B) Sasa-LGA1 and in HKL stimulated with 500 U rIFNa (dark grey), 500 U rIFNc (light grey) 500 U rIFNb (white) or 10ng/ml rIFNγ (black) for 24. Gene expression data were normalized against the reference gene EF1α, and log 2-fold changes were calculated using the unstimulated sample (media alone) at the same time point. The data represent values from [n ϵ (4;8)] individuals, with each dot representing cells isolated from an individual fish. The line intersecting the y-axis at 1 represents the unstimulated control that the fold change of the treatments is in relation to. Significant (p < 0.05) upregulation compared to control (media alone) is indicated by (a) and asterisks indicates the strength of significance: **p < 0.01, ***p < 0.001, and ****p < 0.0001 among the indicted bars. The data presented is representative of two separate experiments.
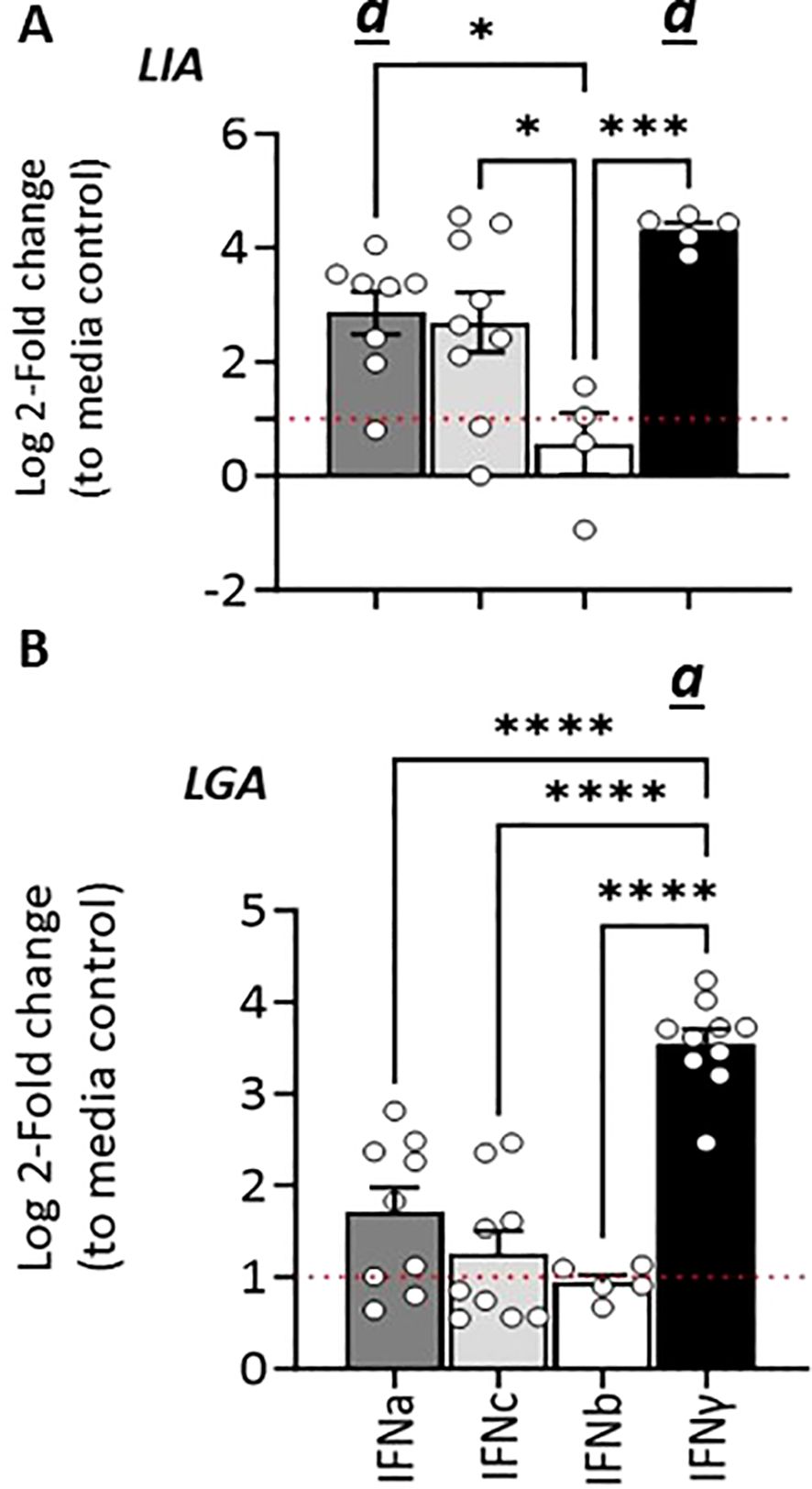
Figure 7. Stimulation with type I and type II IFNs differentially regulates Sasa-/Onts-LIA and Sasa-LGA1 transcription in (A) SSP9 and (B) CHSE-214 cells. Expression of MHC class I L lineage Sasa-LIA/Onts-LIA and Sasa-LGA1 in SSP9 and CHSE-214 cells 24 h post stimulation with 500U rIFNa (dark grey), 500U rIFNc (light grey) and 10ng/ml rIFNγ (black). Gene expression data at each time point were normalized against the reference gene EF1α and fold changes were calculated using control cells stimulated with media alone collected at the same time point. The line intersecting the y-axis at 1 represents the control that the fold change of the treatments is in relation to. Significant (p < 0.05) upregulation compared to the corresponding control group is indicated by (a) and Asterisks indicates the strength of significance between the indicated time points and are indicated by *p < 0.05, ***p = 0.0001, ****p < 0.0001. The data presented is representative of four separate experiments.
Similarly, in SSP-9 cells stimulated with rIFNγ we observed a rapid and marked upregulation of Sasa-LIA expression, a > 20-fold increase in gene expression was observed within 3 hps that peaked (>90-fold) at 24 h post-treatment and then declined somewhat at the last time point analyzed (72 h post-treatment, Figure 7A). Similarly, rIFNa1, and to a lesser extent rIFNc stimulation upregulated Sasa-LIA expression, albeit at a significantly lower level compared to rIFNγ. These results were largely recapitulated in CHSE-214 cells where we observed marked induction of Onts-LIA in response to stimulation with both rIFNγ and rIFNa1 but not to rIFNc (Figure 7C).
In SSP-9 cells the highest level of Sasa-LGA1 induced expression, with a >10-fold induction compared to unstimulated cells, was observed in response to rIFNγ stimulation while rIFNc stimulation resulted in peak of Sasa-LGA1 expression at 6 hps that declined by the 24 h time-point (Figure 7B).
3.4 Pharmacological inhibition of the JAK/STAT pathway reduces rIFNa1, rIFNc and rIFNγ-induced Sasa-LIA and Sasa-LGA1 lineage expressionTo further assess the impact of type I and type II IFN stimulation on L lineage gene induction, we analyzed Sasa-LIA and Sasa-LGA1 expression in SSP-9 cells in the presence or absence of a JAK inhibitor I (a reversible ATP-competitive inhibitor of Janus protein tyrosine kinases (JAKs)). As expected, inhibiting the JAK/STAT pathway resulted in a reduction in IFN type I induced Mx1/2 and IFNγ induced Mx8 gene expression and at a 10-fold higher dose the IFNγ-induced Mx protein levels were visibly affected (Figures 8A-C). Figures 8D, E shows that Sasa-LGA1 and Sasa-LIA transcript levels significantly decreased in rIFNγ -treated cells in the presence of the inhibitor compared to control cells. Reduction in Sasa-LIA transcript levels in the presence of the inhibitor were also apparent in rIFNa1-treated and, albeit not significant, in SSP-9 cells stimulated with rIFNc. Notably, no significant effect of the inhibitor was observed with regard to the basal expression of either Sasa-LIA or Sasa-LGA1 (Supplementary Figure 6).
留言 (0)