
記住我

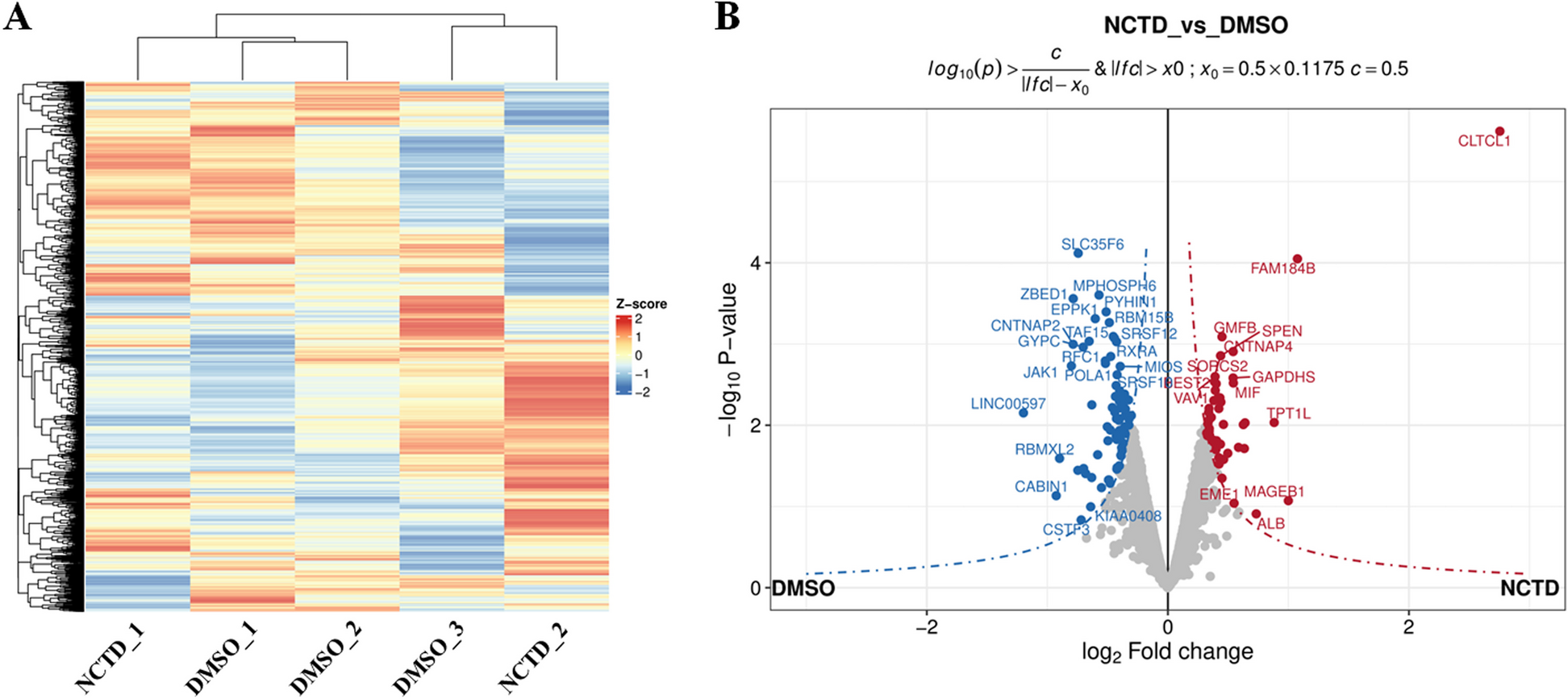





Figure 1 provides a clear step-by-step analysis, outlining the transition from initial data exploration to model construction. In this study, we started with an initial dataset of 4425 proteins and identified 285 DEPs. Simultaneously, we identified 947 metabolism-related proteins and intersected these with the 4425 proteins, resulting in 380 proteins. To identify key proteins linked to the prognosis of polycystic ovary syndrome (PCOS), we conducted univariate Cox regression analysis followed by LASSO regression, which ultimately identified 10 prognostic proteins. These prognostic proteins, combined with clinical data, were used to develop predictive models. The performance of the models was evaluated using ROC curves, DCA, calibration curves, and nomograms.
Fig. 1
illustrates the workflow of proteomics data analysis
Participant clinical characteristicsTable 1 provides an overview of the clinical characteristics of PCOS patients and control subjects. The PCOS group exhibited significantly higher BMI, FINS, and HOMA-IR compared to the control group, indicating greater metabolic disturbances in PCOS patients. The PCOS group also showed trends of higher cholesterol (CHO) and triglycerides (TG) levels, along with lower high-density lipoprotein (HDL) levels, although these differences were not statistically significant. Moreover, the rate of pregnancy loss was higher in the PCOS group compared to the control group, highlighting the increased reproductive challenges faced by women with PCOS. These reproductive issues may be linked to both metabolic disturbances and underlying endometrial dysfunction. Furthermore, the lower live birth rate observed in the PCOS group emphasizes the need for targeted interventions to improve reproductive outcomes in these patients.
Table 1 Participant clinical characteristics of patients with PCOS and controlsProteomic data analysis and screen differential expression proteinsFrom the initial dataset of 4425 proteins, we examined the raw data distribution and applied normalization to ensure uniformity across samples (Fig. 2A-B). The boxplots clearly show that normalization reduces variability and aligns the data distribution more closely between the control and PCOS groups. Principal Component Analysis (PCA) was conducted to illustrate the overall variance in the data and to distinguish between the control and PCOS groups. As shown in Fig. 2C, the PCA plot illustrates a distinct separation between the two groups. Finally, 285 differentially DEPs were identified between the PCOS and control groups, as illustrated by the heatmap in Fig. 2D and the volcano plot in Fig. 2E. The heatmap shows clustering of the DEPs, with distinct expression patterns between the two groups, supporting the findings from PCA that highlight the biological differences between PCOS and control samples. The volcano plot further visualizes the significant upregulated and downregulated proteins, which are critical to understanding the underlying metabolic processes in PCOS.
Fig. 2
Differential Expression Analysis of Proteomic Data. (A) Boxplot of proteomic data before normalization. The black boxes represent the control group, and the red boxes represent the PCOS group. (B) Boxplot of proteomic data after normalization, which showing more uniform distribution. (C) Principal Component Analysis (PCA) plot of the proteomic data, demonstrating the separation between the control group (blue) and the PCOS group (red). (D) Heatmap of DEPs between the control and PCOS groups. The red and blue colors indicate higher and lower expression levels, respectively. (E) Volcano plot of DEPs, with the x-axis representing the log2 fold change and the y-axis representing the -log10 p-value. Red and blue dots represent significantly upregulated and downregulated proteins, respectively, while green dots highlight the most significantly different proteins
Differentially expressed proteins functional enrichment analysisTo further explore the biological significance of the DEPs in PCOS, we performed GO and KEGG pathway enrichment analyses (Supplementary Table e2). GO enrichment analysis of downregulated proteins (Fig. 3A) indicated significant involvement in macromolecule metabolic processes, cellular nitrogen compound metabolic processes, and the regulation of metabolic processes. These findings suggest a suppression of essential metabolic functions in PCOS, potentially contributing to impaired cellular homeostasis and energy production. On the other hand, GO analysis of upregulated proteins (Fig. 3B) indicated significant enrichment in small molecule metabolic processes, lipid metabolic processes, and transporter activity. This suggests that certain metabolic pathways, particularly those related to lipid and small molecule metabolism, are activated in PCOS, possibly contributing to the dysregulated lipid profile commonly observed in these patients. The KEGG pathway enrichment analysis (Fig. 3C) provided further insights into the functional pathways involved. Upregulated proteins were significantly enriched in pathways such as metabolic pathways, glucagon signaling, and glycolysis/gluconeogenesis, indicating a shift towards increased energy mobilization and metabolic dysfunction. Downregulated proteins were involved in pathways such as spliceosome, axon guidance, nucleocytoplasmic transport, and RNA degradation. The downregulation of these pathways suggests a disruption in cellular communication and RNA processing in PCOS, which may further exacerbate reproductive and metabolic disturbances.
Fig. 3
GO and KEGG Enrichment Analysis of Differentially Expressed Proteins. (A) Gene Ontology (GO) enrichment analysis of downregulated proteins. The size of the bubbles represents the count of proteins, and the color gradient indicates the adjusted p-value. (B) GO enrichment analysis of upregulated proteins. (C) KEGG pathway enrichment analysis of differentially expressed proteins. The bar plot displays significantly enriched pathways, with blue bars representing downregulated proteins and red bars representing upregulated proteins. The x-axis representing the -log10 (p-value)
Identification and analysis of prognostic metabolism-related proteinsUnivariate Cox regression analysis, followed by LASSO regression (Fig. 4A-C), identified 20 candidate metabolism-related proteins significantly associated with reproductive outcomes, which were further refined to 10 key prognostic proteins: ACSL5, ANPEP, CYB5R3, ENOPH1, GLS, GLUD1, LDHB, PLCD1, PYCR2, and PYCR3. These proteins were considered to have the most substantial association with reproductive outcomes and were selected for further analysis. To explore the functional relationships among these 10 key prognostic proteins, a protein-protein interaction (PPI) network was constructed using the STRING database (Fig. 4D). The network reveals multiple interactions among the selected proteins, suggesting their involvement in interconnected metabolic pathways. Figure 4E shows the correlation network of the identified metabolism-related DEPs. This network visualizes the correlations based on expression levels. Strong positive correlations may indicate cooperative roles in metabolic processes, whereas negative correlations suggest opposing regulatory effects.
Fig. 4
Identification and Analysis of Prognostic Metabolism-Related Proteins. (A) Forest plot showing the Univariate Cox regression analysis of metabolism-related proteins. The x-axis represents the hazard ratio, and the y-axis lists the proteins with significant associations. The x-axis represents the hazard ratio, while the y-axis lists the proteins that show significant associations with the outcome. (B) Partial likelihood deviance plot from the LASSO regression model, displaying the tuning parameter (lambda) selection process. (C) LASSO coefficient profiles of the metabolism-related proteins. (D) The protein-protein interaction network of these potential Metabolism-related Proteins. (E) The expressions of screened Metabolism DEPs were used to establish a correlated network, with blue lines indicating negative correlations and red lines indicating positive correlations. The thickness of the lines represents the strength of the correlations
Construct the risk prognostic modelWe constructed the risk prognostic model using the ten metabolism-related proteins previously screened. As a result of prognostic data and ten metabolism-related proteins, the risk prognostic signature was developed as follows: Risk score = 0.52 ×expr (ACSL5) -0.28×expr (ANPEP) -0.25×expr (CYB5R3) + 0.11×expr (ENOPH1) -0.94×expr (GLS) -0.10×expr (GLUD1) + 0.47×expr (LDHB) + 0.03×expr (PLCD1)-0.06×expr (PYCR2)-0.36×expr (PYCR3). The heatmap illustrates the expression levels of the selected metabolism-related proteins in high-risk and low-risk PCOS groups. This visualization confirms distinct expression patterns between the two groups, with specific proteins being markedly upregulated or downregulated (Fig. 5A). This differential expression suggests that these metabolism-related proteins may play critical roles in modulating the risk of adverse reproductive outcomes in PCOS. The risk score distribution plot categorizes patients into high-risk and low-risk groups based on their risk scores (Fig. 5B). Figure 5C illustrated the survival status plot correlates risk scores with pregnancy outcomes, showing a clear distinction between live birth and pregnancy loss. The clear separation between the two groups emphasizes the effectiveness of the risk score in predicting reproductive success, highlighting the clinical utility of the prognostic model. Expression levels of the 10 selected proteins were compared between high-risk and low-risk PCOS groups. Boxplots showing the expression differences of 10 selected proteins between high-risk and low-risk PCOS group (Fig. 5D). The Kaplan-Meier survival curve demonstrates a significant difference in survival probability between high-risk and low-risk groups, with the high-risk group having a markedly lower survival probability (Fig. 5E). This indicates the strong prognostic value of the identified metabolism-related proteins in predicting adverse outcomes in PCOS.
Fig. 5
Construct the Risk Prognostic Model. (A) Heatmap showing the expression levels of selected metabolism-related proteins in high-risk and low-risk PCOS groups. Red indicates higher expression, and green indicates lower expression. (B) Risk score distribution plot, with high-risk patients represented by red dots and low-risk patients by green dots. (C) Survival status plot, showing the relationship between risk scores and pregnancy outcomes. Red dots represent pregnancy loss, and green dots represent live birth. (D) The boxplots of the expression levels of the ten significant proteins between high-risk and low-risk groups. (E) Kaplan-Meier survival curve comparing the high-risk and low-risk groups. The y-axis represents survival probability, and the x-axis shows time in weeks. The red line indicates the high-risk group, and the blue line indicates the low-risk group
Evaluation of the risk prognostic modelTo evaluate the performance of our prognostic model, we assessed its ability to predict pregnancy outcomes at different time points: 6, 28, and 37 weeks. The ROC curves for these predictions demonstrated excellent performance, with AUC values of 0.988 at 6 weeks, and perfect scores of 1.000 at both 28 and 37 weeks (Fig. 6A). The model’s predictive performance was compared to traditional clinical variables, including age, BMI, AMH, HOMA-IR, and lipid profiles (Fig. 6B). The protein-based model significantly outperformed all individual clinical markers, with an AUC of 1.000 compared to AUC values ranging from 0.419 to 0.667 for the clinical features. This highlights the superiority of the protein-based model in accurately predicting live birth outcomes, demonstrating its potential as a more reliable tool for risk assessment in PCOS compared to conventional clinical indicators. A nomogram integrating the 10 proteins was developed to predict live birth probability at 6, 28, and 37 weeks (Fig. 6C). The nomogram offers a practical and individualized risk prediction tool that can be utilized in clinical settings, providing a user-friendly way to estimate the likelihood of successful pregnancy outcomes based on protein expression levels. Figure 6D presents the decision curve analysis (DCA) for 37 weeks live birth prediction, showing a clear net benefit for using the protein-based risk model across a range of threshold probabilities. The calibration curve (Fig. 6E) confirmed the accuracy of the model in predicting live birth probability at 37 weeks, with observed outcomes closely matching the predicted probabilities.
Fig. 6
Prognostic Analysis of Selected Metabolism-Related Proteins. (A) Receiver operating characteristic (ROC) curves for predicting outcomes at 6 weeks, 28 weeks, and 37 weeks using the 10 selected proteins. The area under the curve (AUC) values are shown for each time point. (B) ROC curves comparing the predictive performance of clinical data and the selected proteins. (C) Nomogram for predicting the probability of live birth at 6 weeks, 28 weeks, and 37 weeks based on the 10 selected proteins. (D) Decision curve analysis (DCA) for predicting 37-week outcomes, showing the net benefit of the risk prediction model. (E) Calibration curve for predicting 37-week live birth probability, comparing predicted probabilities with observed outcomes
Correlation analysis between clinical data and prognostic proteinsThe correlation analysis was visualized in a heatmap (Fig. 7A), highlighting the relationships between clinical variables (such as BMI, age, and serum lipid levels) and the expression levels of the prognostic proteins. This suggests that higher BMI is associated with reduced GLS expression, potentially indicating metabolic dysregulation linked to impaired glutamine metabolism in individuals with higher body weight. BMI exhibited a significant negative correlation with the expression of GLS (r = -0.44, p = 0.01), as depicted in the scatter plot (Fig. 7B). Additionally, CHO showed a significant positive correlation with the expression of LDHB (r = 0.35, p = 0.04), as shown in Fig. 7C. Elevated LDHB levels in individuals with higher cholesterol could reflect an increased reliance on anaerobic glycolysis, which might be associated with metabolic stress or lipid dysregulation in PCOS patients.
Fig. 7
The correlation between clinical data and prognostic proteins. (A) Heatmap showing the correlation between clinical data and prognostic proteins identified from the study. Pearson’s correlation coefficients are presented, with significant correlations (p < 0.05) marked by *. (B) Scatter plot illustrating the significant negative correlation between BMI and GLS, with a fitted regression line (r = -0.44, p = 0.01). (C) Scatter plot showing the significant positive correlation between CHO and LDHB, with a fitted regression line (r = 0.35, p = 0.04)
留言 (0)