
記住我





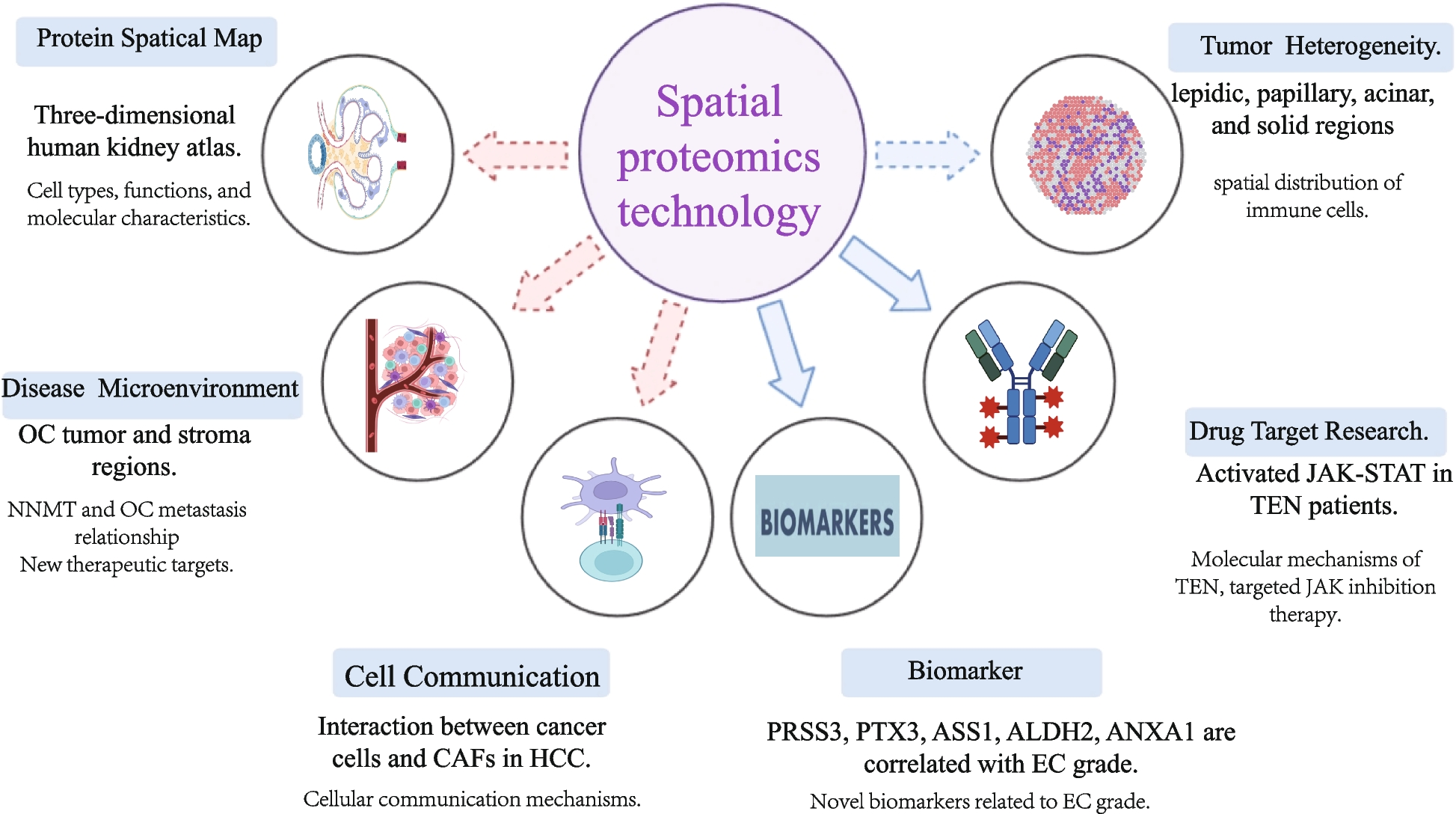

Spatial proteomics is an innovative approach that combines mass spectrometry with advanced imaging techniques, allowing researchers to accurately obtain high-resolution localization information of proteins within cells. Subcellular spatial proteomics has previously been summarized. For example, Lundberg et al. extensively covered methods and applications, encompassing mass spectrometry-based organelle analysis, protein–protein interaction networks, and whole-cell imaging of protein localization, highlighting the link between abnormal subcellular localization and diseases. Other researchers have reviewed various methods for subcellular protein localization, such as fluorescence imaging, protein proximity labeling, organelle purification, and mass spectrometry, emphasizing the importance of protein localization in cellular signaling, growth, proliferation, movement, and programmed cell death (see detailed techniques in references [10, 11]). Subcellular-level proteomics research has been utilized in cystic fibrosis (CF) studies. For instance, John J et al. analyzed the transport pathways of CFTR protein in CF, revealing that golgin proteins are involved in the aberrant transport of CFTR, which contributes to the disease's molecular pathology [12]. Meanwhile, Hirst et al. focused on the critical role of AP-5 in the transport process from late endosomes to the Golgi apparatus. Additionally, studies have shown that VX-809 (lumacaftor) induces extensive mitochondrial remodeling, facilitating CFTR functional restoration [13, 14]. These studies enhance the understanding of CF's molecular pathology and facilitate the development of new diagnostic and therapeutic targets. Given this, the review will focus on the introduction of spatial proteomics at the tissue level, building on the solid foundation provided by subcellular spatial protein analysis.
In gene expression research, although mRNA levels are typically considered direct indicators of gene activity, the correlation between mRNA and protein abundance is often low, failing to accurately reflect actual protein abundance. This lack of correlation arises from various mechanisms: mRNA molecules are prone to degradation due to regulation by RNA-binding proteins and miRNAs, and translation efficiency can limit protein synthesis [15, 16]. Moreover, post-translational modifications and protein transport barriers or mislocalization, as well as protein degradation pathways like the ubiquitin–proteasome system, further affect protein function and abundance [17, 18]. Thus, measuring mRNA levels alone cannot fully reflect the actual distribution and functional state of proteins. Spatial proteomics not only allows for the direct monitoring and quantification of protein changes and modifications but also provides new perspectives and methods for discovering disease-related protein biomarkers and understanding the complex structure of the human proteome in disease contexts.
Traditional proteomics primarily focuses on the compositional structure and function of proteins, reflecting the overall protein expression changes in an organism through identification and quantitative analysis. In contrast, spatial proteomics offers higher spatial resolution and comprehensiveness, allowing direct analysis of protein localization and interactions [19]. This aids in comprehensively understanding disease mechanisms and discovering new protein biomarkers and drug targets. Spatial proteomics enables high-throughput quantitative and localization analysis of proteins in histological samples. Depending on the quantification method, current spatial detection techniques for proteins are mainly divided into three categories: fluorescence-based antibody, mass spectrometry-based, and sequencing-based spatial proteomics methods.
Fluorescence-based antibody spatial proteomics methodsFluorescence-based antibody spatial proteomics methods employ specific antibodies or fluorescent probes to label target proteins, ensuring precise protein localization. Fluorescence microscopy is then used to observe and record the spatial distribution of proteins at the tissue and cellular levels, offering high resolution. Immunohistochemistry was the earliest method in this category, detecting protein expression in tissue sections using specific antibodies. Its advantage lies in preserving the original tissue structure of proteins. However, a limitation of this traditional approach is that it requires precise antibody matching and can only localize one protein per experiment. To increase the diversity of protein detection and overcome the limitations of immunohistochemistry, some research groups have introduced multiplex immunofluorescence techniques. This method extends protein detection diversity by using multiple fluorescent probes, allowing simultaneous detection of up to 100 protein biomarkers in a single tissue section, significantly enhancing high-throughput analysis capabilities [20]. Nevertheless, it encounters challenges of spectral overlap with different fluorescence signals, limiting its applicability for quantitative analysis of multiple proteins in complex samples. Tyramide Signal Amplification (TSA), a popular multiplex immunofluorescence technique, uses tyramide enzyme to enhance the fluorescent signal of target proteins, improving protein sensitivity [21]. Increasing signal intensity facilitates the detection of low-abundance proteins but introduces potential issues of cross-reactivity or non-specific binding, requiring longer multiple staining cycles. This method is suitable for diverse labeling or localization of low-abundance proteins. The development of sequential immunofluorescence (seqIF) has addressed these challenges. This technique automates the application of antibodies and their elution in a continuous process, combining integrated microscopy with microfluidic chips for in situ imaging, optimizing sample processing workflows and providing a reliable method for detecting multiple biomarkers [22]. It features efficient antibody incubation and washing capabilities, yet its experimental procedures are complex, suitable for high-resolution localization and interaction studies of proteins in tissue or cell culture samples. One of the most representative imaging-based spatial proteomics methods is CODEX, which primarily uses antibody-conjugated DNA barcodes, multiple rounds of staining and elution, and imaging systems to identify barcode sequences, achieving high-throughput detection of multiple proteins in samples. The method allows simultaneous detection of multiple high-resolution markers, with a detection limit of 50 proteins [23, 24]. It is applicable to various sample types such as fresh and fixed tissues (e.g., paraffin sections), but its dependence on antibodies limits its application to a broader range of markers (Table 1). It is well-suited for highly multiplexed biomarker detection and spatial distribution analysis at the single-cell level, particularly in exploring tumor microenvironments.
Table 1 Summary of key characteristics of major spatial proteomics techniquesMass spectrometry-based spatial proteomics methodsAs mass spectrometry technology has rapidly evolved, it has become a mainstream technique for protein analysis. It identifies proteins based on the sequence of their unique peptides. The principle involves extracting proteins from tissue samples and subjecting them to proteolytic digestion with trypsin, followed by separation of the protein peptides using a mass spectrometer. The analysis of mass spectrometric signals by the spectrometer and image analysis software reveals changes in protein expression. Spatial proteomics methods based on mass spectrometry are divided into spatially untargeted and targeted proteomics.
Non-targeted mass spectrometry methods maximize proteome coverage, commonly used for biomarker development [31]. Liquid chromatography-mass spectrometry (LC–MS) was one of the earliest spatially untargeted methods, combining the advantages of liquid chromatography and mass spectrometry for the separation and purification of proteins in mixtures [26]. LC–MS can analyze complex biological samples such as blood, urine, and cell lysates with high sensitivity in studying protein quantification, post-translational modifications, and protein interactions. It is suitable for research on disease biomarkers and drug development. However, limitations in automation and complex sample preprocessing restrict its widespread application in clinical disease research. In contrast, Mass Spectrometry Imaging (MSI) is a relatively fast and cost-effective method that, as a key tool combining mass spectrometry and imaging technologies, allows for label-free acquisition of data on metabolites, lipids, peptides, and proteins from tissue slices. MSI is suitable for fresh or frozen tissue sections, requiring minimal sample preparation and using various ionization techniques to measure hundreds of potential analytes' relative spatial abundance in almost any tissue section [32, 33]. Sample preparation for MSI is relatively simple, with high performance, but limited quantitative capabilities. It is ideal for unbiased spatial distribution analysis at the tissue level, such as protein expression patterns in cancer tissues or drug distribution in tissues. Secondary ion mass spectrometry (SIMS), the first MS imaging technology applied in the 1960s, is suitable for solid samples like tissue sections or material surfaces. SIMS achieves extremely high spatial resolution by bombarding the sample surface with an ion beam and analyzing the generated mass spectrometry signals. However, its ionization capability for proteins limits its application in complex protein samples [34]. SIMS is suitable for high-resolution imaging analysis of protein expression in specific regions or cell types but requires highly pure samples and clean surfaces. The most typical imaging mass spectrometry technique introduced thereafter was Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging (MALDI-MSI). MALDI-MSI typically combines with time-of-flight (TOF) mass spectrometry. It suits various sample types. requiring uniform matrix coating on tissue, followed by laser ablation of the solid matrix and analytes from tissue sections, with TOF analyzers deducing the mass-to-charge ratio of desorbed ions to determine ion mass [25]. Although the spatial resolution of MALDI-MSI, ranging from 5–50 µm [25, 35], is lower compared to SIMS technology, and the choice of matrix significantly influences ionization results, presenting challenges in detecting low-abundance proteins, it is characterized by high throughput, capable of detecting 50–100 peptides without destroying tissue. Therefore, this method is suitable for quantifying the spatial distribution of multiple molecules and biomarkers in tissue sections.It has been widely applied in quantifying and detecting various biomarkers in tissues, serum, plasma, cerebrospinal fluid, and urine [36, 37]. Desorption Electrospray Ionization Mass Spectrometry Imaging (DESI-MSI) technology, which developed subsequently, uses an electrospray ion source to generate ions on the surface of liquid samples [38], allowing direct molecular information retrieval from tissue surfaces without the need for matrix selection, thus avoiding the matrix effects observed in MALDI-MSI. DESI-MSI is suitable for various sample types with simple preparation. However, it has lower spatial resolution and is used in studies that require rapid screening of large-scale samples or do not necessitate high-resolution information. Although MALDI-MSI and DESI-MSI can unbiasedly analyze the spatial distribution of molecules within tissues, they also have limitations. The lack of predefined targets increases the difficulty of identifying peptides in the vast amount of data. High-efficiency chromatographic separation techniques can effectively separate complex samples and reduce matrix effects, thereby enhancing resolution. The absence of chromatographic pre-separation in the aforementioned methods results in complex and overlapping signals in mass spectrometry data, where low-abundance peptides are often overshadowed by high-abundance signals, limiting the capacity for in-depth analysis. Additionally, sample heterogeneity and variations in ionization efficiency pose challenges for accurate quantitative analysis. To overcome these challenges, laser capture microdissection (LCM) technology has emerged. LCM is an advanced tissue separation technology that combines laser cutting and image analysis, allowing for the rapid, precise selection and localization of regions of interest (ROI) from tissue samples. This one-step collection avoids sample contamination from manual handling and achieves separation at the single-cell level without losing spatial information [39, 40]. Laser Capture Microdissection-Mass Spectrometry (LCM-MS) is the most widely applied method. It involves processing and mass spectrometry analysis of fixed, stained tissue sections, correlating spatial information with data through image analysis software to identify protein expression and interactions within tissues [27] (Fig. 2).
Fig. 2
Operational procedure of spatial proteomics mass spectrometry methods. 1. Sample Collection: Different tissue samples are collected. 2. Slice Staining: Tissue samples were sectioned and mounted onto glass slides. Following natural air-drying, appropriate staining techniques, such as Hematoxylin and Eosin (H&E) staining, immunohistochemical staining, Masson’s trichrome staining, etc., were chosen based on experimental design. 3. Laser Fiber cutting: The laser microdissection system is used for precise localization and microdissectionof the regions of interest under a microscope. 4. Protein Extraction: were extracted using protease or protein extraction buffer.. 5. Protein Digestion: The extracted proteins were subjected to digestion to release peptide fragments. 6. LC–MS/MS: The peptide solution is injected into the liquid chromatography system, where separation of different peptide segments is achieved using a chromatographic column. Subsequently, the separated peptide segments are introduced into the mass spectrometer, where they are ionized. The mass spectrometer evaluates the protein mass and relative abundance by analyzing the mass-to-charge ratio of ions in the peptide segments. 7. Raw Data: The data can be preprocessed through baseline correction, peak fitting, and other methods. 8. Statistical Analysis: The utilization of data visualization tools for differential analysis on raw data allows for the revelation of information concerning the distribution and abundance of specific proteins across different spatial context
Accurate analysis of protein expression in tissue samples is a key challenge in proteomics research, where LCM-MS has made significant advances, making it suitable for studying specific tissue separations. With the continuous development and expansion of LCM-MS's analysis capabilities, researchers can now achieve higher throughput in protein analysis. For example, a novel high-throughput protein mapping method combines voxelization techniques with high-throughput LC-FTICR mass spectrometry to analyze smaller tissue units in the mouse brain, successfully generating expression patterns for 1,028 proteins. This technique utilizes stable isotope labeling and AMT tag strategies to enhance the reliability and reproducibility of the analysis. However, further improvements in resolution are needed [41]. Min Ma et al. proposed the Micro-scaffold Assisted Spatial Proteomics (MASP) strategy, utilizing 3D-printed micro-scaffolds to achieve higher spatial resolution. They mapped over 5,000 proteins in the mouse brain, achieving uniform micro-compartmentalization and precise spatial information retention across the entire tissue. Although this requires more complex analysis procedures, the technique significantly enhances sample processing efficiency and proteomics analysis capability for small samples [42]. The development of NanoPOTS technology has enabled researchers to perform grid-based sampling at a 100 µm resolution on mouse uterine tissue sections, achieving automated mass spectrometry imaging of 2,000 proteins. This technique significantly improves upon LCM-MS, particularly through the use of microfluidic sample processing and automation, which reduces sample loss and enhances sensitivity and throughput [43]. Integrating LCM, MSI, and spatial perception algorithms, the research team revealed the spatial heterogeneity of protein expression and tumor-associated markers in human atypical teratoid/rhabdoid tumors. This approach further enhanced the identification of protein spatial expression and provided a more in-depth characterization of tissue heterogeneity [44].
Spatial targeted mass spectrometry methods play crucial roles in current research for verifying pre-determined proteins. Key techniques include Imaging Mass Cytometry (IMC) and Multiplexed Ion Beam Imaging (MIBI). IMC involves staining tissue with metal-conjugated antibodies, followed by scanning the tissue with a laser to excite the metal ions and generate mass-to-charge signals, which are analyzed to localize and quantify proteins. It is suitable for high-dimensional spatial resolution studies like tumor microenvironment analysis, offering high multiplexing capability and robustness but with complex sample preparation and high costs [28]. MIBI, a variant of IMC, is based on secondary ion mass spectrometry analysis, distinguishing different antibody markers through ion beam-excited secondary ion signals [29]. It excels in high-resolution spatial analysis and can simultaneously analyze up to 100 targets, overcoming the resolution limitations of MALDI-MSI. It is ideal for highly multiplexed and high-resolution analysis of complex biological samples. However, the high cost of metal tags and long acquisition periods limit its widespread application [45, 46].These methods still require improvements in tissue sample preparation and accuracy. Sample preparation issues like tissue washing and protease digestion may cause protein delocalization, affecting spatial resolution and accuracy. Recent research has proposed several improvements in sample preparation and data processing, such as Filter-Aided Sample Preparation (FASP), Single-Pot, Solid-Phase-enhanced Sample Preparation (SP3), and optimized MS data acquisition strategies [47,48,49,50]. These methods significantly improve sample preparation efficiency and data reliability, broadening mass spectrometry's application scope.
Sequencing-based spatial proteomics methodsDigital Spatial Profiling (DSP) is an emerging sequencing-based spatial proteomics and transcriptomics technology, providing a means for highly multiplexed analysis at the tissue section level. Suitable for tissue sections or cultured samples, DSP involves fixation and sectioning to ensure sample quality and efficiency. DSP's principle involves in situ binding of probe-tagged antibodies to tissue samples, selecting regions of interest (ROIs) through UV light exposure to separate probe tags from bound proteins, and collecting tags in a 96-well plate, each well corresponding to an ROI. Ncounter system quantifies probe tags in wells to determine protein expression abundance. DSP's advantages include high spatial targeting, allowing for multiple immunofluorescence pretreatments of tissue sections, selecting ROIs, and strong multiplexing capabilities and high sensitivity, which make it adaptable to diverse sample types. Nevertheless, the technique's dependency on antibodies and the risk of signal overlap could potentially affect the reliability of the results. DSP is widely used in various tissue sample studies, particularly in cancer, immune, and neurological disease research. For example, Cabrita et al. used DSP to study melanoma patient samples, analyzing molecular characteristics of tertiary lymphoid structures formed by tumor-associated CD8 + T cells and CD20 + B cells, identifying TLS-related molecules predictive of survival outcomes [51]. Another example involved DSP analysis of tumor and stroma regions in pancreatic cancer patient tissues, identifying key markers related to treatment, providing important clues for biomarker screening [30].
留言 (0)