Cell lines
The HAP1 cells are a near-haploid human cell line derived from the KBM-7 chronic myelogenous leukemia (CML) cell line (Horizon Cat #C631). The HAP1 Parental (Control) and NEK10 KO (Knockout) haploid cells were produced by CRISPR/Cas9 and obtained from New Horizon/Perkin (line HZGHC002995c011). The NEK10 gene was edited by CRISPR/Cas9 by New Horizon/Perkin Elmer. This specific region is a highly conserved nucleotide sequence among the NEK10 isoforms. The target transcript is NM_199347.
The Guide RNA Sequence are: TGCTTGATTGGACGTTCAAA;
PCR Forward: CAGAGAAACAGCCAGATGAAGAAAG;
PCR Reverse: GTGCTTACTGAAACATCTGGTCATT.
The NEK10 KO cells have a 35 bp (base pair) removal in the NEK10 KO gene compared with control cells (Parental) (see Supplemental Fig.S1).
For cell maintenance, Dulbecco's modified IMDM medium was used in the presence of 10% fetal bovine serum and antibiotics [1 ml fungizone (amphotericin, cat#15,290–018) and 300 µl Gibco gentamicin (cat#15,750–060) per 500 ml of media]. For HAP1 cells, in our experiments, we considered early cell passages less or equal to 25 cell passages and late cell passages more than 25 cell passages.
The human HeLa cell line was obtained from the ATCC and maintained as recommended. For cells maintenance, was used Dulbecco's modified DMEM medium in the presence of 10% fetal bovine serum and antibiotics [1 ml fungizone and 300 µl gentamicin per 500 ml of media].
For NEK10 knockdown we used a lentiviral system of short interfering RNAs (shRNAs): (shNEK89: 5'-CATTGCCAGAACACATTATAT-3'; shNEK90: 5'GCCTCGTCCAGATATTGTAGAA-3'). The pLKO.1 empty vector was used as a control (pLKO.1) and the shRNAs were obtained from the RNAi Consortium (TRC, IRB-Barcelona, Spain). Lentiviruses carrying shRNAs were produced and collected at the Laboratory of Viral Vectors (LVV, LNBio / CNPEM-Campinas, SP, Brazil). Lentiviruses were transduced in HeLa cells in the presence of 1 μg/ml of polybrene and a complete medium for 24 h. For selection, was used puromycin (Sigma-Aldrich, St. Louis, MO, USA) at 3 µg/ml.
Oligonucleotides and Probes
For NEK10 knockout test—NEK10 F2 5'- TGTTATTCTCTCTCCTAGGG—3'; NEK10 F1 5'-CATTGTGTTATTCTCTCTCC -3'; NEK10 B2 5'- AGA-TATGTTAAATGGAGCCC-3'; NEK10 B1 5'- GAGCCCTTTGGAAACATAT—3'.
For Mycoplasma test – MycoFoward 5'-GTAATACATAGGTCGCAAGCGTTATC-3'; MycoReverse5'-CACCATCTGTCACTCTGTTAACCTC-3'.
For TaqMan-real time PCR of mtDNA quantification- ND1 Forward 5’-GAAGTCACCCTAGCCATCATTC-3’; ND1 Reverse 5′-GCAGGAGTAATCAGAGGTGTTC -3′; Probe ND15′-/5TET/AAGGGTGGA/ZEN/GAGGTTAAAGGAGCC/3IABkFQ/-3. COI Forward 5′-TTCTGACTCTTACCTCCCTCTC-3′; COI Reverse 5′-TGGGAGTAGTTCCCTGCTAA-3′; Probe COI 5′-/56FAM/TCCTACTCC/ZEN/TGCTCGCATCTGCTA/3IABkFQ/-3′. Actin Forward 5′-GTCACCGGAGTCCATCAC-3′; Actin Reverse 5′-GCCATGTACGTTGCTATCCA-3′, Probe Actin 5′-/5Cy5/TGCCAGTGGTACGGCCAGAG/3IAbRQSp/-3 [15].
Antibodies
Western blotting: Anti-NEK10 goat polyclonal (D-17; Santa Cruz Biotechnology cat#sc-103067); rabbit anti-ATAD3B (1:1000 Proteintech 16,610–1-A); mouse anti-total phosphothreonine (1:1000 Cell Signaling (42H4) 8954); mouse anti-total phospho serine Arg-X-Tyr/Phe-X-pSer (1:1000 Cell Signaling 2981); rabbit anti-total phospho Tyrosine (1:1000 Cell Signaling P-Tyr-1000 8954S); rabbit anti-HSP60 (1:1000 Cell signaling D6F1); mouse anti-TOM20 (1:1000) Santa Cruz SC17764; mouse anti-tubulin A (1:1000 Protein Tech 66,031–1); rabbit Anti-AIF (1:1000 Cell Signaling D39D2); mouse anti-complex III UQCRC2 (1:1000 Abcam ab14745); mouse anti-complex I NDUFA9 (1:1000 Abcam ab14713); mouse anti-SDHA (1:1000 Abcam ab 14,715); mouse anti-ATP5A (1:1000 Abcam abI5H4C4).
Immunoprecipitation: rabbit anti-TOM20 (1:2000 Cell Signaling D39D2); rabbit anti-HSP60 (1:2000 Cell signaling D6F1); rabbit anti-ATAD3B (1:2000 Proteintech 16,610–1-A).
Immunocytochemistry: rabbit anti-TOM20 (1:200 Cell Signaling D39D2); rabbit Anti-AIF (1:200 D39D2 Cell Signaling); Alexa Fluor anti-rabbit 488 (1:250 Invitrogen).
OXPHOS Reagents
Oligomycin, FCCP, antimycin A, and rotenone were supplied by the Seahorse OCR assay kit. Sodium pyruvate, glutamine, and glucose were obtained from Sigma/Aldrich.
Fluorescent Dyes
MitoBright Deep Red- MitoBright LT dyes MT10 (Dojindo Bio) are designed to be retained in the mitochondria for long-term visualization and it is not dependent on a strong membrane potential. TMRM—The MitoProbe TMRM Assay Kit M20036 contains tetramethylrhodamine methyl ester for detecting the mitochondrial membrane potential state (Thermofisher Scientific Inc.). MitoSox—MitoSOX Red super-oxide indicator M36008 (Thermofisher Scientific Inc.)
Zeocin Treatment
Cells were treated with 300 µg/ml zeocin for 3 h in culture medium, followed by 3 h recovery.
Genotyping the HAP1 NEK10 knockout cells by PCR
For each reaction, was added 5 μl of 10 × reaction buffer (Green Reaction Buffer Thermo Scientific); 20 ng of plasmid DNA; 1 µl of each oligonucleotide (10 µM); 1 µl dNTP (10 mM); 0.5 μl of Dream Taq polymerase (Thermo Scientific) and volume adjusted to 50 μl with MilliQ water. For the PCR reaction, the following parameters were used: 1 cycle of 95ºC for 1 min, 33 cycles of 95ºC for 30 s; 55 °C for 30 s; and 72ºC for 1 min/ plasmid kb, and finally, 1 cycle of 72ºC for 15 min. PCR products were analyzed is a 12% polyacrylamide DNA gel, electrophoresed in TBE buffer (Tris/Borate/EDTA, pH 8.0).
Mitochondrial respiration analysis
For HAP1 cells (Parental and NEK10 KO), we used the Seahorse XFp Analyzer (Agilent, CA), which measures extracellular O2 through fluorescence sensors. Cells were plated in XFp Seahorse-specific plates. The phenol-free Seahorse XF medium was supplemented with 10 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate. Inhibitors were used at the following concentration: 1 µM oligomycin; 1.5 µM FCCP; 0.5 µM antimycin/rotenone. The results were analyzed on Seahorse Wave Desktop Software 2.6.1.
For HeLa cells (pLKO, sh89, and sh90), was used the Seahorse XF24 Analyzer (Agilent, CA). Cells were plated in XF24 Seahorse-specific plates. DMEM medium without phenol was supplemented with 25 mM glucose and 4 mM glutamine. Inhibitors were used at the following concentration: 1 µM oligomycin; 500 nM FCCP; and 1 µM antimycin/rotenone for mitochondrial respiratory analyses. The results were analyzed on Seahorse Wave Desktop Software 2.6.1. Quantification of OCR values subtracted non-mitochondrial oxygen consumption values.
Transmission Electron Microscopy (TEM)
The HAP1 cells (Parental and NEK10 KO) or HeLa cells (pLKO, sh89, and sh90) cells were plated in 6-well plates. On the next day, cells were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate and 3 mM CaCl2 buffer for 5 min at room temperature, followed by a 1 h incubation on ice. After fixation, the samples were washed three times in 0.1 M sodium cacodylate and 3 mM CaCl2 buffer and post-fixed with 1% osmium tetroxide with 0.8% potassium ferrocyanide in 0.1 M sodium cacodylate buffer (pH 7.4), for 30 min, and then block stained with 2% uranyl acetate on ice-cold overnight.
The cells were dehydrated with ethanol at 4ºC and infiltrated in Epon resin. After four changes of resin solution, a fifth change of resin was performed and immediately placed in a vacuum oven at 60 ◦C to be polymerized for 72 h.
Ultrathin sections were cut with a Leica Ultracut microtome, stained with 2% uranyl acetate and Reynolds lead citrate, and examined in a Tecnai G2 Spirit BioTWIN (FEI Company, Hillsboro, OR, USA). Supplemental figures were obtained with a LEO 906-Zeiss transmission electron microscope at an accelerating voltage of 60 kV. TEM was performed at the Electron Microscopy Laboratory of the Institute of Biology at the University of Campinas. Mitochondrial morphology analysis was performed using Image J2 software. Cristae number was quantified as the number cristae segments spanning the matrix. The statistical analyses were performed with GraphPad Prism, and t-test or one-way ANOVA depending on the number of groups. Tukey's test was employed for post hoc analysis.
Immunocytochemistry
Parental and NEK10 KO cells were plated in 24-well plates with coverslips previously sterilized with UVC. When the cells reached 70% confluence, immunocytochemistry was performed. The cells were fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were washed with PBS 2 × rapidly. After that, the cells were permeabilized with 0.1% Triton X-100 and again, washed with PBS for 2 × quickly. Cells were blocked with 2% BSA for 10 min at room temperature. The coverslips with cells were incubated with a primary antibody (anti-AIF or anti TOM20) diluted in 0.2% BSA for 45 min at room temperature in a humid chamber. The cells were washed twice with PBS for 5 min, each wash. The cells were incubated with a specific secondary antibody for immunofluorescence diluted in 0.2% BSA for 45 min at room temperature in a humid chamber. Cells were washed twice with PBS for 5 min each. Coverslips were overlaid on slides with EverBriteTM Mounting Media with DAPI (Biotium Cat#23,004). The AIF immunofluorescence intensity was obtained by the Image J2 Software.
Flow Cytometry
For flow cytometry analysis, Parental and NEK10 KO cells were plated in 6-well plates one day before. On the day of the procedure, cells were treated or untreated (assay negative control). For mitochondrial membrane potential analysis was used 20 nM MitoProbe TMRM. For mitochondrial mass analysis was used 0.1 µmol/l MitoBright Deep Red. For mitochondrial superoxide, analysis was used 5 µM MitoSox and 20 µM Antimycin A. Cells were washed 1 × with PBS, trypsinized, and the cell pellets were washed 2 × with PBS. Cells were resuspended in 400 µl PBS with 2% fetal bovine serum and analyzed on the BD FACSAria TM Ilu Special Order System. For each analysis, were collected 50.000 events. The data were analyzed with the software FCS Express 7 at the Flow Cytometry Core at the University of Miami, Miller School of Medicine.
Real-Time qPCR
To determine the total levels of mtDNA present in the samples, we performed qPCR using TaqMan reagents (PrimeTime Std qPCR Assay, Integrated DNA Technologies). Samples were analyzed on the BioRad CFX96/C1000 qPCR instrument. The comparative cycle threshold (Ct) method was used to determine relative values, and total mtDNA levels were determined by comparing mtDNA ND1 (TET) or COX1 (FAM) with genomic DNA actin (Cy5). For the real-time cycle, the following protocol was used: 95º. C for 3 min at 95°. C for 0.15 min and 60°. C for 1 min for 39 cycles [15].
Crude Mitochondrial Fractionation
Parental and NEK10 KO cells in different passages were plated in six 175 cm2 flasks four days before the crude mitochondrial fractionation assay. On the day of the procedure, the cells were washed twice with PBS (1 × Phosphate Buffered Saline—pH 7.4) and trypsinized. The pellet was washed twice with 1 × chilled PBS, and the cells were kept at 4 °C. The pellet was resuspended in T-K-Mg solution [10 mM Tris–HCl pH 7.0; 10 mM KCl; 0.15 mM ice-cold MgCl2] containing protease inhibitors (cOmplete™ Protease Inhibitor Cocktail Sigma/Roche®) and phosphatase inhibitors (PhosStop Sigma/Roche®) and left on ice for 5 min. Cells were homogenized with 60 strokes in a mechanical homogenizer for cell lysis. Sucrose and Tris–HCl pH 7.0 were added to the homogenate for a final concentration of 0.25 M and 10 mM, respectively. Cells were centrifuged at 1500 g for 3 min at 4 °C to obtain the nucleic fraction. The supernatant was centrifuged at 8000 g for 10 min at 4ºC to obtain the cytoplasmic fraction (supernatant) and the mitochondrial fraction (pellet). Depending on the procedure, the mitochondrial fraction was resuspended in solution A [20 mM Tris HCl p7.2; 0.25 M sucrose; 40 mM KCl; 2 mM EDTA] to be kept at -80º C or lysed with RIPA buffer [30 mM HEPES, pH 7.4,150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 5 mM EDTA, (cOmplete™ Protease Inhibitor Cocktail Sigma/Roche®) and phosphatase inhibitors (PhosStop Sigma/Roche®)] for LC–MS/MS experiments, or lysed with Cell Signaling Lysis Buffer for immunoprecipitation assays.
DNA Extraction
Parental and NEK10 KO cells were grown in T-25 flasks for two days, washed 2 × with PBS, trypsinized, and centrifuged at 1000 × g for 10 min, and the pellet was used for DNA extraction. The NucleoSpin Tissue XS kit from Macharey-Nagel Ref # 740,901.250 was used. The DNA extraction protocol was followed according to the manufacturer's specifications.
Mycoplasma test
Parental and NEK10 KO cells were grown in T-25 flasks for 5 days with antibiotic-free IMDM medium, washed 2 × with PBS, trypsinized, and centrifuged at 1000 × g for 10 min and the pellet was used for DNA extraction. After DNA extraction, the PCR reaction described below. For each reaction was added 1 μl of 10 × reaction buffer (Green Reaction Buffer Thermo Scientific); 0.5 μl of each oligonucleotide (Myco F and R 10 μM); 1.6 µl of dNTP (10 mM); 0.1 μl of Dream Taq polymerase (Thermo Scientific) and the volume adjusted to 10 μl with MilliQ water. The following parameters were used: 1 cycle of 95ºC for 5 min, 32 cycles of 95ºC for 30 s; 65 °C for 1 min; and 72 °C for 1 min/ plasmid kb, and finally, 1 cycle of 72 °C for 5 min.
Analysis of Liquid Chromatography—Mass Spectrometry (LC-M/MS)
The protocol below was used at the Proteomics Core of the University of Florida in Jupiter, FL, USA. The statistical analysis and graphs were performed at the University of Miami.
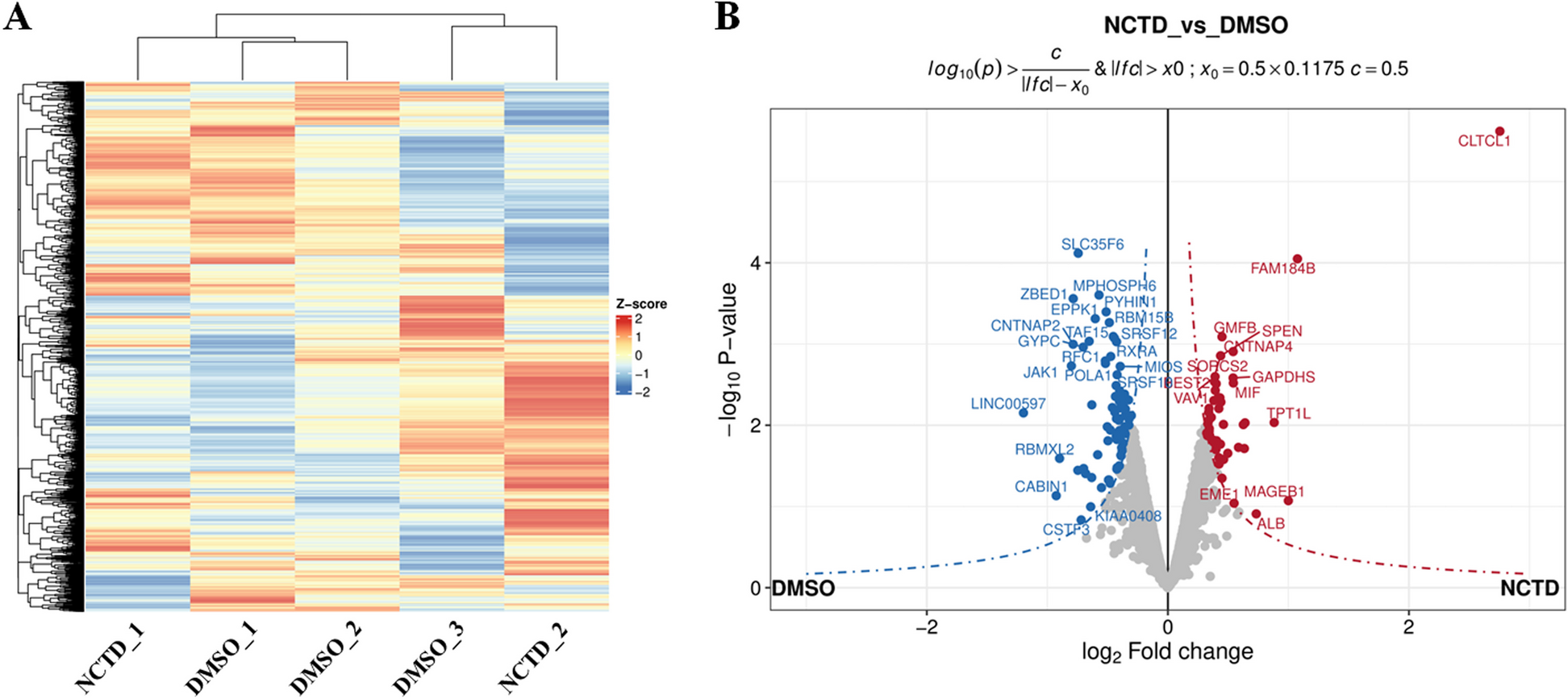
Quantitative analysis of the TMT pro-6plex experiment was performed simultaneously with protein identification using Proteome Discoverer 2.5 (PD) software. The precursor and fragment ion mass tolerances were set to 10 ppm, and 0.6 Da, respectively); the enzyme was Trypsin with a maximum of 2 missed cleavages and FASTA files for Uniprot Human (UP000005640, downloaded on 9/14/2020) proteome, and common contaminants were used in SEQUEST searches. The impurity correction factors obtained from Thermo Fisher Scientific for the TMT 6plex kit were included in the search and quantification. The following settings were used to search the non-enriched sample data; dynamic modifications; Oxidation / + 15.995 Da (M), Deamidated / + 0.984 Da (N, Q), N-Terminal modification of Acetyl / + 42.011 Da (N-Terminus), Met-loss / -131.040 Da (M), Met-loss + Acetyl / -89.030 Da (M) and static modifications of TMTpro / + 304.207 Da (Any N-Terminus, K) and MMTS + 45.988 Da (C). Only unique + Razor peptides were considered for quantification purposes. The percolator feature of PD was used to set a false discovery rate (FDR) of 0.01. The samples were normalized using the “Total Peptide Amount” option at PD2.5. The protein Abundance Based method was used to calculate the protein level ratios. Co-isolation threshold and SPS Mass Matches threshold were set to 50 and 65, respectively.
The following settings were used to search the phospho-enriched data; dynamic modifications; Oxidation / + 15.995 Da (M), Deamidated / + 0.984 Da (N, Q), Phospho / + 79.966 Da (S, T, Y), N-Terminal modification of Acetyl / + 42.011 Da (N-Terminus), Met-loss / -131.040 Da (M), Met-loss + Acetyl / -89.030 Da (M) and static modifications of TMT6plex / + 229.163 Da (N-Terminus, K), MMTS / + 45.988 Da (C). Only unique + Razor peptides were considered for quantification purposes. The percolator feature of Proteome Discoverer 2.5 was used to set a false discovery rate (FDR) of 0.01. IMP-ptmRS node (Taus et al. 2011) was used to calculate probability values for each putative phosphorylation site. The “Total Peptide Amount” normalization method was used to adjust for loading bias, and non-phosphorylated peptides were excluded from normalization and ratio calculation. The protein Abundance Based method was used to calculate the protein level ratios. Co-isolation and SPS Mass Matches thresholds were set to 50 and 0, respectively.
At the peptide level, peptide isoforms were defined by the Peptide Isoform Grouper node that summarized PSMs that share the same sequence and modifications. Phosphopeptide isoforms (PI) were further evaluated by removing not quantified or not phosphorylated PIs. At the peptide and protein levels, phospho-enriched and non-enriched samples were analyzed using the following statistical method. Contaminant proteins/peptides were excluded from the data set, and signal/noise values were normalized using log2 transformation. Statistical analysis was performed using paired T-Test with GraphPad Prism version 9.5 (Graph Pad Software, San Diego, CA, USA). Proteins changes with p-value < 0.05 were considered significant.
Immunoprecipitation
To perform immunoprecipitation, the mitochondrial pellet was lysed with cell lysis buffer (Cell Signaling®) containing protease inhibitors (cOmplete™ Protease Inhibitor Cocktail Sigma/Roche®) and phosphatase inhibitors (PhosStop Sigma/Roche®). The mitochondrial pellet was incubated with a specific primary antibody at a dilution of 1:2000. After 1 h on the shaking platform at 4 °C, G-Sepharose was added, followed by 1 h on the shaking platform at 4 °C. The G-Sepharose-Ac-protein complexes were obtained by centrifugation, washed with lysis buffer for 5x, and the proteins extracted with Laemmli buffer and analyzed by Western Blotting.
Western Blotting
Protein concentrations were measured with Bio Rad's DC Protein Assay reagent. Proteins were added to 10% Mini-PROTEAN® TGX™ Precast Protein Gels, 10-well, 50 µl #456,103, and transferred to Trans-Blot Turbo Mini 0.2 µm PVDF Transfer Packs #1,704,156, the transfer was carried out in Trans-Blot Turbo Transfer System. Immunoblot analysis was performed with primary antibodies diluted in a blocking solution (2% bovine serum albumin and 0.02% sodium azide in PBS-buffered saline). Protein bands were detected with peroxidase-conjugated antibodies and visualized by ECL chemiluminescence (Amersham). The images were acquired with ChemiDoc MP Imaging System. The intensity of the bands were obtained by the Image J2 Software [16].
Pathway analyses
The pathway analyses (Reactome and GO [Gene Ontology] Biological Processes) of phosphoproteins and non-phosphoproteins were performed using the online tool Enrichr 27 [17] available at https://maayanlab.cloud/Enrichr. Enrichr analyses gene sets in several ontology databases.
Statistical analysis
Statistical analysis was performed using One-Way ANOVA, followed by Tukey's or Dunnett’s Multiple Comparison Test, or the paired or unpaired T-Test. GraphPad Prism version 9.5 (Graph Pad Software, San Diego, CA, USA) was used for statistical analysis and graph design. The statistical analysis is shown in Mean with SD or with SEM and represented by * when it is considered significant, where p-value < 0.05 = *, p-value < 0.01 = **, p-value < 0.001 ***, and p-value < 0.0001 = ****.







留言 (0)