
記住我
The equine encephalitis complex, including western, eastern, and Venezuelan equine encephalitis viruses (WEEV, EEEV, and VEEV), are New World alphaviruses in the family Togaviridae (Aguilar et al., 2011; Calisher, 1994). These viruses are collectively referred to as equine encephalitic viruses (EEVs) throughout this manuscript. These viruses are maintained in an enzootic cycle between mosquitoes and an animal host (e.g., rodents and/or birds) but can spill over into both humans and horses, causing illness (Ronca et al., 2016). In horses, mules, and donkey's, equine encephalitis can cause appetite loss, flu-like symptoms, and progress to muscle and nervous system degeneration (Guzmán-Terán et al., 2020). In humans, illness can include flu-like symptoms and can also progress to neurological deficits and seizures. These effects occur most commonly in children for whom EEVs are more likely to cause life-long behavioral deficits, seizures, and delirium (Falchek, 2012). Cases that progress to neurological deficits, including encephalitis require significant long-term care, which was estimated to cost $320,000 per individual in 1971, and $400,000 per year per individual in 1995 (Villari et al., 1995; Earnest et al., 1971). Based on the 2.54% inflation of the economy between 1995 and 2023 (Webster, 2024), this equates to ~$790,000 per year in 2023. Despite over 100 years of research on these viruses, there are no FDA-approved antiviral treatments or vaccines against EEV infection in humans. Ultimately, current treatment options are limited to supportive care. The lack of FDA approved therapeutics for EEVs is due to a variety of challenges, including the limited number of laboratories researching these viruses because of biosafety level limitations and the lack of adequate and standardized animal models.
1.2 TBITraumatic brain injury (TBI) is a leading cause of death and disability across the world, with an estimated 69 million people sustaining an injury each year (Dewan et al., 2018; Peterson et al., 2022). The CDC defines a TBI as “an injury that affects how the brain works” and may be caused by a “bump, blow, or jolt to the head, or a penetrating injury” (CDC, 2023a). Following the primary insult, “secondary injury” will occur in the minutes, hours, days, weeks, and years' post-injury. Secondary injury leads to chronic neurological deficits due to cell death, inflammation, and other consequences of primary injury. Additionally, TBI is a risk factor for several neurodegenerative disorders and neurological disorders and impairments (Dams-O'Connor et al., 2016; Delic et al., 2020; Rapoport, 2012; Howlett et al., 2022). Both primary and secondary injury lead to acute seizures in 1/5 individuals who receive a TBI and chronic post-traumatic epilepsy (PTE) occurs in ~1/50 cases (Fordington and Manford, 2020). Due to the heterogeneous nature of the TBI mechanism, pathophysiology, severity, and outcome, developing effective therapeutic strategies and treatments has yielded limited success, with no correct or universal FDA-approved treatments available (Kochanek et al., 2020; Nishimura et al., 2022). Several recent review articles and reports cover new technological advances in TBI research (Bowman et al., 2022; Bonanno et al., 2022; Lu et al., 2015; Ahmed, 2022). These focus on developments in imaging, biomarkers, and therapeutic approaches that offer insights into the pathophysiology and treatment of TBI. For instance, innovations in high-density neurophysiology monitoring systems, alongside advanced neuroimaging and bioinformatics, have enabled more precise tracking of brain injury progression and recovery. Technologies like high-resolution MRI, PET imaging, and blood-based biomarkers are helping researchers characterize injury severity and predict outcomes more accurately. Therefore, novel approaches to studying TBI and its related pathophysiology may enable therapeutic development.
1.3 NAsOrganophosphate compounds (OPs) are primarily divided into nerve agent OPs and pesticide OPs. Exposure to OPs causes the death of 300,000 people per year worldwide (Ahmad et al., 2024; Adeyinka et al., 2024), primarily from occupational exposure to pesticide OPs and exposures in underdeveloped countries. Challenges in identification of agent in a warfare setting make estimation of deadly nerve agent OP exposures difficult and likely underestimated (Costanzi et al., 2018; Gunnell et al., 2007) though there are recent high-profile examples of nerve agent OP use on civilians (Chai et al., 2017; Haslam et al., 2022; John et al., 2018; Morita et al., 1995). For this review we will focus on nerve agents (NAs) as these have significant implications as warfare agents (Mukherjee and Gupta, 2020). NAs irreversibly inhibit acetylcholinesterase activity, causing a buildup of acetylcholine in both the central and peripheral nervous systems that produces significant neurological changes (Costanzi et al., 2018). NAs are typically broken into classes, including the “German” G-series agents sarin (GB), soman (GD), tabun (GA), and the Russian “Venomous, Victory, or Viscous” V-series agents, which consists of VE, VG, VM, VR, and VX. Nerve agent mechanisms of action are consistent across the series, but the volatility and toxicity vary. V-series nerve agents, most notably VX, are less volatile at ambient temperature and generally regarded as more toxic than G-series agents, but this toxicity is typically associated with skin contact and persistent environmental hazard (Jang et al., 2015; Wiener and Hoffman, 2004; Rosenblatt et al., 1996). G-series agents are volatile at room temperature leading to more significant toxicity through inhalation or skin contact. An additional class, the Novichoks and A-series agents, will not be discussed in this review as their classification, chemical structures, and mechanisms are not well characterized by the literature. The little information that is present concerning this series is incomplete and from scrutinized sources, whereas G-series and V-series agents are relatively well characterized in literature. A-series and Novichok agents in particular display an extreme level of toxicity not observed in other nerve agents (Opravil et al., 2023; Noga and Jurowski, 2023).
Exposure to NAs can occur through inhalation, ingestion, or skin absorption (Wiercinski and Jackson, 2024). Small amounts of these chemicals can cause a variety of mild to moderate flu-like symptoms such as nausea, vomiting, confusion, headache, and weakness or other cholinergic symptoms such as watering eyes, drooling, blurred vision, increased heart rate, muscle spasms, and sweating (CDC, 2023b). The onset of symptoms is typically rapid at high doses, but long-term low dosage exposure can induce memory impairment, motor dysfunction, depression, and anxiety, with multiple lines of evidence from Tokyo Subway Attack victims, veterans, and exposed Iraqi civilians (Figueiredo et al., 2018). The mechanism by which long-term low-dose exposure of NA induces depression and behavioral changes is not well understood: however, neuronal damage and degeneration is commonly correlated with cognitive and motor dysfunction observed post-mortem (Figueiredo et al., 2018). Lower doses of NAs also can cause convulsion mediated at peripheral neuromuscular junctions, high levels of NA exposure almost invariably result in prolonged or repetitive central seizures (status epilepticus, SE) or death without immediate medical intervention (Hrvat and Kovarik, 2020). Unique to NAs, most long-term neurological changes are due to SE-induced neuronal damage, inflammation, and changes to neuronal signaling (Figueiredo et al., 2018) as NAs do not have directly toxic effects on neural cells (Aroniadou-Anderjaska et al., 2023).
2 Neuropathology in humans and laboratory animal models 2.1 EEVsHuman case studies of VEEV, EEEV, and WEEV are extensively under-documented due to the initial symptoms often presenting as febrile illness as well as non-specific documentation due to these viral illnesses being classified under a larger umbrella of neuroinvasive viruses, including flaviviruses, West Nile virus (WNV), and Japanese encephalitis virus (JEV) (Calisher, 1994; Ronca et al., 2016). The number of reported VEEV, WEEV, and EEEV cases trace back to the 1930s, the same time frame in which these viruses were isolated and mosquito vectors were determined to be the route of transmission (Weaver et al., 2004a; Zacks and Paessler, 2010). The combined impact of WEEV, VEEV, and EEEV includes >300,000 cases, >300 recorded deaths, and 3,000 survivors with long-lasting neurological disorders, including paralysis, seizures, migraines, and depression (Crosby and Crespo, 2024) (Table 1). As previously mentioned, this is likely an underestimate due to many viral infections causing vague symptoms in humans, and therefore these infections may go undiagnosed and untested. A recent study identified elevated glial fibrillary acidic protein (GFAP) in patient serum from acute VEEV and Madariaga virus (MADV) infections; however this biomarker was not specific to alphavirus infection as bacterial cases with encephalitis also displayed elevated GFAP (Bartlett et al., 2024). EEV pathology in humans has been divided into three distinct phases consisting of a lymphotropic phase (early/short term), a neuroinvasive phase, and a neurodegenerative phase (late/long-term) [reviewed in Steele and Twenhafel (2010); Kehn-Hall and Bradfute (2022)]. Neurological complications can be attributed to both the neuroinvasion and replication of EEVs in the brain, as well as from the inflammatory response. Despite the large range of neurological deficits induced by these viruses, there is little known about the mechanism of neuroinvasion in humans. Similarly, it is debated whether the inflammatory process hallmarked by the increase of neutrophils in the central nervous system (CNS) is harmful or helpful (Peiseler and Kubes, 2019; Drescher and Bai, 2013). Therefore, we rely upon animal models of infection to study pathologic, behavioral, and molecular processes altered by infection and ultimately identifying therapeutics. Animal models of EEVs have previously been reviewed (Ronca et al., 2016; Steele and Twenhafel, 2010; Kehn-Hall and Bradfute, 2022); however, here, we seek to highlight the consequences of neuroinvasion and long-term neurological symptoms of EEVs in order to correlate them across different neurological diseases.
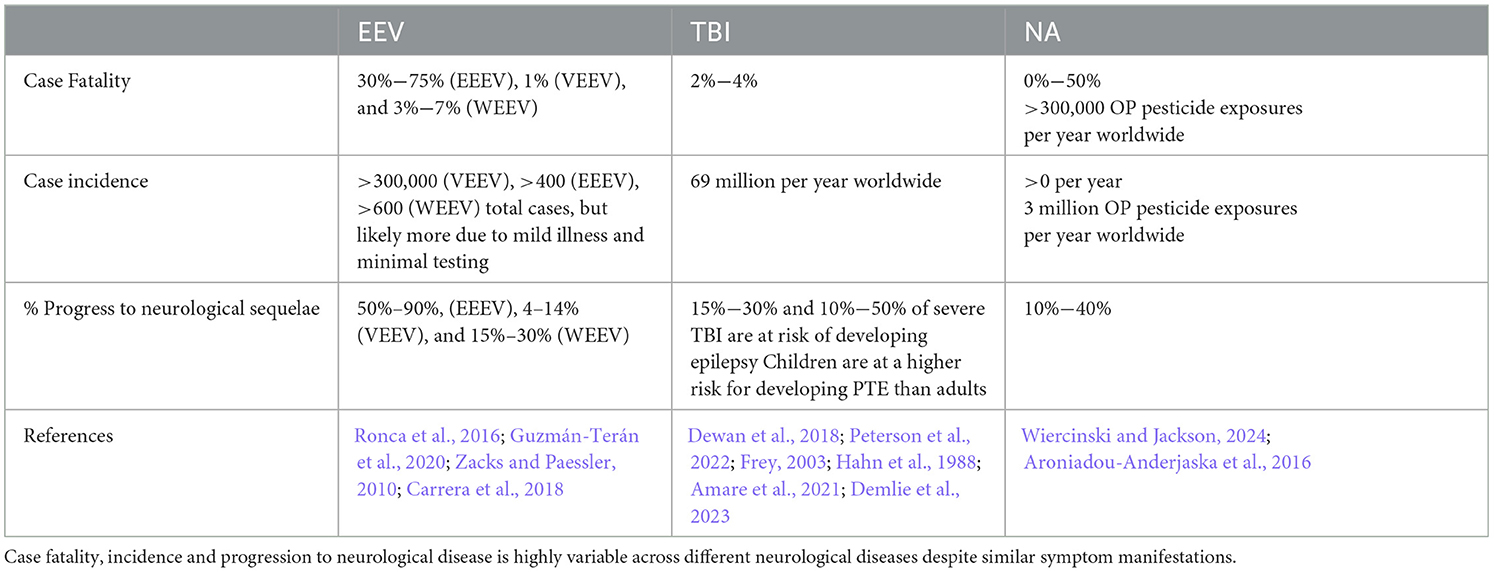
Table 1. Impacts of EEVs, TBI, and NA on humans.
Animal models used for EEV research include non-human primates, rats, gerbils, mice, guinea pigs, rabbits, and hamsters, which have been extensively reviewed by others (Steele and Twenhafel, 2010; Kehn-Hall and Bradfute, 2022; Dremov and Solianik, 1977). Of these models, Guinea pigs, hamsters, and rabbits have high fatality rates, including severe lymphoid necrosis prior to invasion into the CNS; therefore, they are less ideal models for studying neurological sequelae. In this review, we focus on rodents (i.e. mice, rats, and some hamster models) and non-human primates (NHPs) as animal models of EEV infection, where neurological infiltration has been well established. It's also important to note that studies in animal models have primarily been reliant upon the utilization of a variety of different strains of VEEV, EEEV, and WEEV, including naturally evolving strains (VEEV Subtypes I-VI, VEEV Trinidad Donkey (TrD), EEEV Georgia Fatal, EEEV FL93-939, EEEV North American, WEEV MacMillan (MCM), WEEV IMP 181) as well as some attenuated viral isolates previously reviewed (e.g. VEEV TC-83) (Steele and Twenhafel, 2010; Sharma and Knollmann-Ritschel, 2019). Route of infection is also important to consider as there are multiple infection methods used including intranasal, aerosolization, and subcutaneous infection; however, there are different degrees of morbidity and mortality across the different infection routes. While EEVs are naturally transmitted via mosquito bite, there is a large interest in understanding pathogenesis that results from inhalational exposure. EEVs are readily aerosolized, VEEV was developed as a biological weapon, and a large number of laboratory acquired infections have occurred via VEEV aerosolization (Rusnak et al., 2018; Weaver et al., 2004b).
2.1.1 VEEVVEEV infections in humans result in a relatively low mortality rate (<1%); however, the progression to neurological deficits is significant with 4%−14% of cases progressing to neurological signs with a higher incidence in children, elderly, and immune compromised people (Lundberg et al., 2017). The two largest outbreaks of VEEV occurred through mosquito transmission in Texas in 1971 and Columbia in 1995 (Aguilar et al., 2011). Collectively, there were >300 deaths reported and 3,000 cases with long-lasting neurological disorders including paralysis, seizures, migraines, and depression (Crosby and Crespo, 2024). Additional symptoms included reduced sensory perception of taste, hearing, and smell and changes in emotional stability and mental fitness (Carrera J. P. et al., 2013; Bowen et al., 1976). These studies highlight significant infection rates and neurological sequelae resulting from VEEV outbreaks.
VEEV infection in NHPs results in clinical symptoms such as weight loss, lethargy, hunching, hyperactivity aggression, photophobia, and full body and partial tremors, which closely resembles infection in humans (Table 2) (Burke et al., 2019). Aerosolized VEEV exposure typically results in prolonged febrile state for 1–8 days post-infection (DPI) with immediate invasion into the brain, whereas subcutaneous administration typically presents as febrile illness for 1–6 DPI with signs of virus in the brain and neurological symptoms such as depression 2–3 DPI (Reed et al., 2004; Weaver et al., 2012; Gleiser et al., 1961; Ludwig et al., 2001). The intranasal route of exposure led to the most severe illness in NHPs with invasion, replication, and lesions due to viral infection as early as 48 h post infection (hpi). Further investigation of infected NHPs identified neuronal necrosis, lesions in the thalamus and olfactory cortex, and perivascular cuffing and gliosis with severe inflammation in the hippocampus and cortex (Reed et al., 2004; Gleiser et al., 1961; Danes et al., 1973; Victor et al., 1956; Smith et al., 2020). In Rhesus Monkeys, vertical transmission via fetal contraction of VEEV was confirmed via intranasal infection of the mother. The fetuses showed viral replication in both the brain and peripheral organs, which led to vision impairment and abnormal brain growth, including microcephaly, hydrocephalus, and porencephaly in 67% of cases (London et al., 1977). Evaluation of VEEV in NHP has several advantages as they display highly similar patterns of febrile illness, lethargy, and depression to human case studies; however, murine models of infection include several cost-effective and higher throughputs benefits not represented in this model.
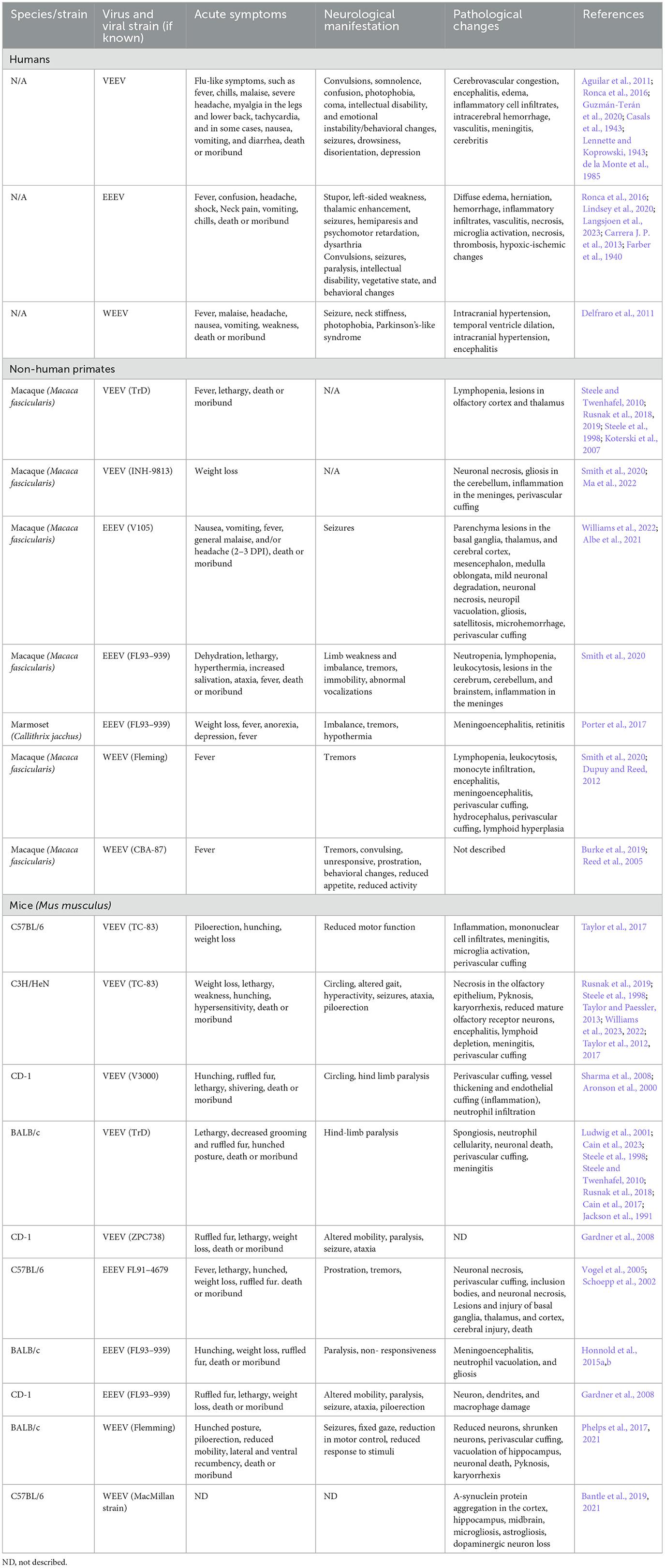
Table 2. Comparison of human EEV cases and laboratory animal models of EEV infection.
Infection in mice via footpad or subcutaneous injection leads to viral replication in the lymphatic system as early as 4 hpi, viremia is detected at 12 hpi, and virus in the brain between 24–72 hpi whereas aerosolization or intranasal infection leads to infection in the brain 16–48 h post-infection (Davis et al., 1994; Rusnak et al., 2019). Multiple strains of mouse models (C57Bl/6, CD-1, BALB/c, C3H/HeN) have been well established with intranasally VEEV-infected mice showing signs of weight loss, tremors, paralysis, and dehydration (Table 2) (Gardner et al., 2008; Berge et al., 1961; Hart et al., 1997, 2000; Vogel et al., 2005; Honnold et al., 2015a,b). In most models of VEEV infection, there is direct entrance into the brain via the olfactory bulb via aerosolization and intranasal routes, which has aided in further understanding the neurological features of infection (Cain et al., 2023; Phillips et al., 2016; Salimi et al., 2020). VEEV can also enter the brain via transcytosis through brain epithelium, independent of blood-brain barrier (BBB) breakdown, which appears later in the course of infection (Salimi et al., 2020). Caveolin-1 was shown to be important for viral neuroinvasion (Salimi et al., 2020). VEEV infection typically results in 100% mortality in mice when they are infected with fully virulent BSL-3 strains of VEEV. However, VEEV TC83 is a live attenuated vaccine strain that is used to study VEEV pathogenesis at BSL-2. VEEV TC83 infection results in different levels of mortality dependent on the strain of mouse utilized. C3H/HeN mice are highly susceptible to VEEV TC83, resulting in 100% mortality when intranasally infected with 109.1 and 107.1 cell culture infection dose 50 (CCID50) (Julander et al., 2008). It's been proposed that higher mortality rates in VEEV-infected C3H/HeN mice compared to C57BL/6 and BALB/c mice is due to a reduced immune response partially dependent on reduced IgA in C3H/HeN mucosa (Hart et al., 1997; Steele et al., 1998; Charles et al., 1997). The majority of C57BL/6 mice infected intranasally with VEEV TC-83 survive infection and serve as an important model to study neurological sequelae following VEEV infection (Ronca et al., 2017; Cain et al., 2017). These mice display biphasic disease initially causing symptoms such as ruffled fur, weight loss, lethargy, and shivering (Figure 1). Following neuroinvasion, circling, hind limb weakness or paralysis, convulsions, and head-tilt are observed. Post-mortem analysis of VEEV V3000 (molecular clone of fully virulent VEEV TrD) infected mice indicated perivascular cuffing, inclusion bodies, neuronal necrosis, and overt encephalitis widespread across the brain (Sharma and Knollmann-Ritschel, 2019; Cain et al., 2023). It has been confirmed that in mice, VEEV primarily infects neurons, but also infects microglia, macrophages, and oligodendrocytes as well as inducing significant inflammation around the BBB (Sharma and Knollmann-Ritschel, 2019).
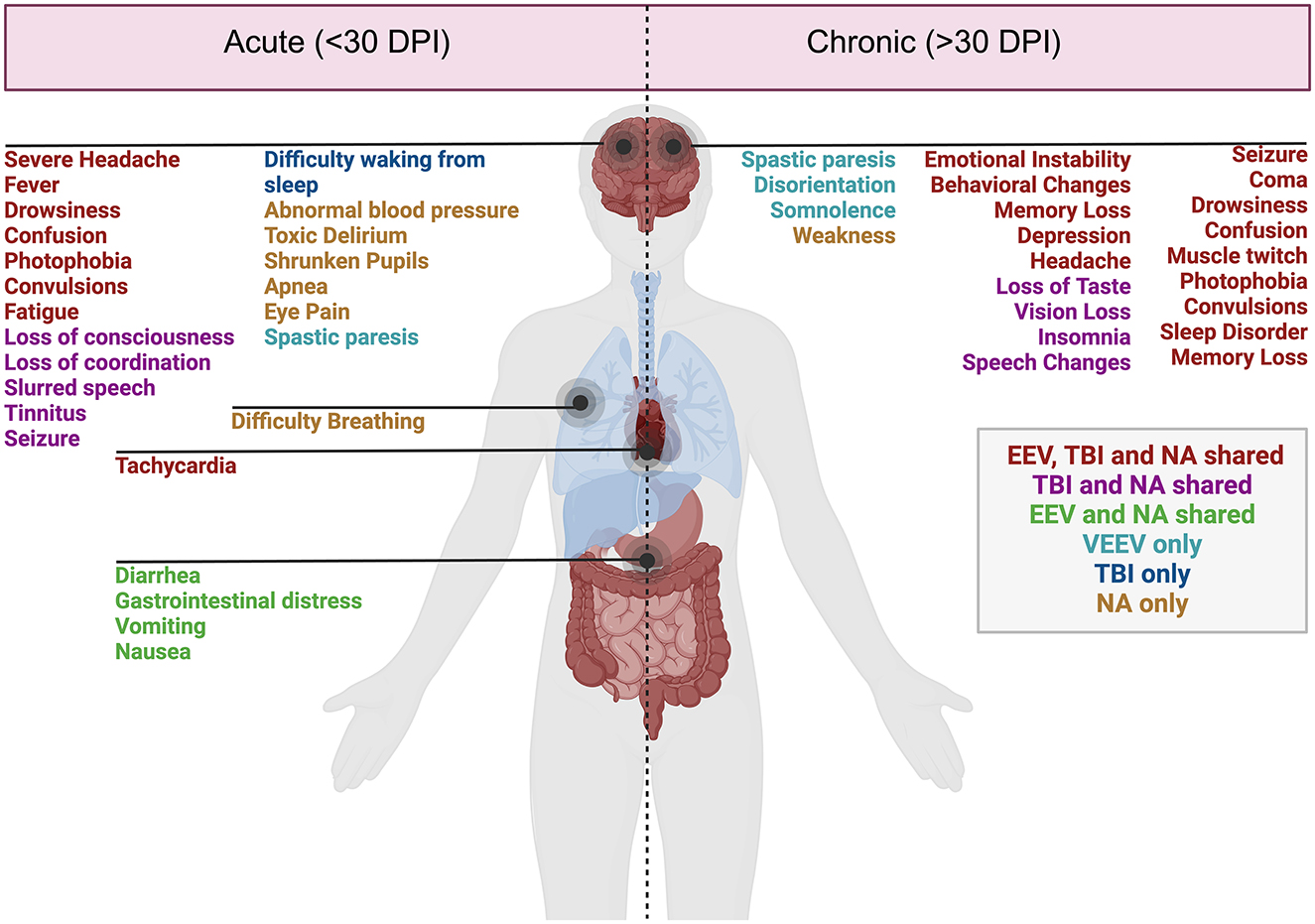
Figure 1. Acute and chronic clinical comparisons between EEVs, TBI, and NA. Summary of characterized acute (<30 days post-injury or exposure) and chronic manifestations (>30 days post-exposure) of disease in humans. Overlapping clinical symptoms and sequelae are color-coded. EEV, TBI, and NA (red), TBI and NA shared (magenta), VEEV and NA shared (green), VEEV only (teal), TBI only (blue), and NA (gold). DPI, days post injury/infection. Created in BioRender. Kehn-hall (2024) BioRender.com/r18b546.
There has been a significant effort to determine transcriptomic changes associated with VEEV infection, although much has been done in cell culture with increasing evaluation in murine models. In VEEV-infected mice, neuronal damage has been correlated with upregulation of apoptotic, antiviral, T-cell response, and pro-inflammatory genes including CXCL10, IL1-beta, Interferon gamma (IFN-y), CCL5 (RANTES), CCL3 (MIP-1a) and TNF-alpha (Weaver et al., 2004a; Sharma and Knollmann-Ritschel, 2019; Sharma et al., 2008; Taylor and Paessler, 2013). Symptomatic mice appear to have more significant upregulation of inflammatory genes, as well as signs of increased astrocyte activation within specific areas of the brain, such as the thalamus and hippocampus, even after 6 months post-infection (Ronca et al., 2016). A more expansive study investigated individual areas of the brain on days 1–7 post-infection and identified upregulation of numerous cell death pathways (phagocytic pathways, natural killer, pyroptosis, necroptosis) and pro-inflammatory response (Williams et al., 2023). Collectively, these studies have established that there is much similarity between humans, NHPs, and mice with CNS invasion leading to markers of encephalitis such as perivascular cuffing, neuronal necrosis, as well as other markers of damage including infiltration of leukocytes, brain lesions, and gliosis. Evaluation of proteomic and metabolomic changes is currently limited to cell culture-based studies of VEEV and is thus not included in this review.
2.1.2 EEEVEEEV infections result in 30%−75% mortality and 50%−90% of surviving individuals progress to long-term neurological symptoms (Lindsey et al., 2020; Ciota, 2022; Langsjoen et al., 2023). Collectively, chronic symptoms are poorly characterized in EEV infected animals due to the high mortality rate. Small periodic cases of EEEV and the emergence of four diverse EEEV lineages from 1960–2010 have yielded inconsistent reporting of morbidity, mortality, and neurological sequelae (Arrigo et al., 2010). There were ~178 documented cases of EEEV infection from 2009–2020 with the majority of these cases in Panama and the eastern United States, with the highest number of cases in Georgia, New Jersey, New York, North Carolina, and Michigan (Carrera J. P. et al., 2013; Virmani et al., 2010; Hill et al., 2023; Vilcarromero et al., 2010). Surveillance efforts identified 100 potential positive cases and seven confirmed cases of EEEV in Panama in 2010, where many survivors experienced chronic seizures which were traced to virally induced abnormalities within the temporal lobe of the brain, responsible for memory storage, formation, sensory processing, and emotions (Carrera J. P. et al., 2013). Sequelae observed in human cases include behavioral, memory, and emotional changes which are potentially attributed to changes such as neuronal loss, vasculitis, thrombosis, gliosis in motor neurons, and abnormalities in the temporal lobe (Table 2) (Ronca et al., 2016; Reddy et al., 2008).
In NHP models of EEEV infection, high fatality rates have been recorded in correlation with the high fatality rates seen in humans (Williams et al., 2022). Studies in NHPs have identified EEEV replication in the CNS and severe neurological disease (Albe et al., 2021; Ma et al., 2022). NHPs infected with EEEV display neuronal dysfunction in the olfactory bulb, olfactory tract, and spinal cord which is associated with EEEV pathology rather than neuronal death, which is also typically seen in VEEV. An overall review of NHP EEEV infections has identified pathological evidence of vasculitis, perivascular cuffing, edema, hemorrhage, and widespread necrosis widespread across the brain (Steele and Twenhafel, 2010; Reed et al., 2004; Arrigo et al., 2010). Another unique characteristic of EEEV in Cynomolgus macaques is the neuroinvasion and pathology in the brain is equally as severe between both intranasal and subcutaneous injection, potentially due to EEEV entering the brain via the bloodstream rather than olfactory nerve routes (Smith et al., 2020).
Mouse models of EEEV are also relatively understudied. EEEV is highly fatal in mice, typically 100% in all routes of infection with accompanying invasion into the brain even with subcutaneous methods as early as 24 h post-infection (Vogel et al., 2005). EEEV infected mice show preferential infection of osteoblasts, skeletal muscle, and fibroblasts, with less efficient replication in macrophages and dendritic cells (Steele and Twenhafel, 2010; Gardner et al., 2008; Vogel et al., 2005). Cytokine analysis indicated CCL5 (RANTES), CXCL9 (MIG), CCL4 (MIP-1B), and IFN-y peak between day 1 and 2 post infection dependent on the route of infection (Honnold et al., 2015a). In CD-1 mice infected with 103 plaque forming units of either VEEV or EEEV, both viruses showed similar trends with decreased weight, piloerection, paralysis, seizure, and 100% mortality by day 7 (Gardner et al., 2008). Disease symptoms of VEEV onset earlier with mice surviving longer with clinical signs of CNS infection; whereas EEEV-infected mice showed later onset of symptoms and mortality within 12–24 h post disease onset (Gardner et al., 2008). Potentially, this is driven by cellular tropism differences between VEEV and EEEV, as EEEV replicates poorly in lymphoid tissues while VEEV flourishes in macrophages and dendritic cells in these tissues (Gardner et al., 2008). The overarching similarity between VEEV and EEEV infections in mice are encephalitis; however, in VEEV-infected mice, neurons in the hippocampus and cerebellum show signs of morphological changes correlated with apoptosis, whereas in EEEV seem to infiltrate the thalamus, pons, and putamen with widespread neuronal necrosis (Vogel et al., 2005). There are a few consistent differences noted with VEEV and EEEV infection in mice which has not been fully elucidated in NHP and humans where paralysis is more common in VEEV and have a rarer occurrence of seizures, while with EEEV there are few cases of paralysis and frequent report of seizures (Gardner et al., 2008). Collectively, EEEV pathogenesis and neuroinvasion appear to be more rapid, although EEEV infected mice display widespread neurological damage and proinflammatory activation similar to VEEV with slight alterations dependent on the route of exposure.
2.1.3 WEEVSeveral hundred cases of WEEV infection have been well documented, with neurological sequelae lingering for several years, if not lifelong, post-infection. In children and some adults, mild sequelae include loss of taste, changes in speech, decreased fine motor skills, changes in gait, and hearing deficits (Table 2) (Mulder et al., 1951). Severe sequelae in both adults and children include personality and behavioral changes, intellectual disability, seizure, limb weakness, mood swings, depression, anxiety and paranoia (Palmer and Finley, 1956; Fulton and Burton, 1953; Deaton et al., 1986). Neurological manifestation of WEEV infections are the most significantly documented partially attributed to its lower mortality (< 7%) and high incidence of neurological symptoms (15%−30%) (Luethy, 2023; Simon et al., 2023). Documentation of WEEV in an aerosol laboratory-acquired infection lead to the death of 2/5 infected individuals following symptoms such as headache, fever, tachycardia, and increased heart rate (Hanson et al., 1967). A recent outbreak of WEEV occurred in early 2024, where >100 equine cases, most of them fatal, and >100 human cases were documented with 10 fatalities (Campos et al., 2024). Campos et al. (2024) identified a novel WEEV lineage likely due to unreported cases circulating in South America, but it does not currently appear to be a recombination with other EEVs. In this outbreak, most cases were mild or asymptomatic, with signs or meningitis and encephalitis in fatal cases (Campos et al., 2024). Interestingly, some patients with WEEV have been described to have other neurological symptoms that mirror those diagnosed with diseases such as Parkinson's and Schizophrenia (Bantle et al., 2019; Levine and Griffin, 1992; Herzon et al., 1957). One recent case occurred in November 2023 of an equine agricultural working in Argentina, where the patient presented with headache, fever, disorientation, confusion, and tiredness, leading to intensive care treatment for 12 days and eventual discharge nearly 30 days after initial symptoms (World Health Organization, 2023). Given both a recent case and severity of disease across many documented cases, it highlights the need for adequate surveillance, diagnosis, and animal models for therapeutic evaluation against WEEV.
WEEV infections in animal models have substantial research in murine, hamster, and NHP models. Studies of WEEV in NHPs have been minimal since the 1930s. Fever and increased heart rate, as well as clinical signs suggesting encephalitis, were observed in NHPs exposed to WEEV via aerosol (Smith et al., 2020; Reed et al., 2005). Some conflicting reports indicate minimal clinical signs and no recoverable virus from WEEV infected NHPs, but potentially this is due to quicker clearance compared to the other EEVs (Burke et al., 2022). Pathology from infected NHPs has indicated viral infection broadly across the brain with widespread infiltration in the gray matter of the brain in areas of the striatum and cerebrum and infection of the neurons in the hippocampus (Reed et al., 2005). Some models of WEEV and VEEV in NHPs suggest peak neutrophil and monocyte infiltration is slightly later around 7–9 DPI. Despite several studies that have investigated WEEV neuropathology, there is still much left unknown about the course of disease in NHPs, highlighting a significant gap in the literature.
Golden hamsters infected with WEEV resulted in 100% mortality with symptoms such as severe respiratory challenges, blurred cornea, eye discharge, and seizure with confirmation of the virus in the brain within 24 h of infection (Zlotnik et al., 1972). Older studies have revealed neuronal necrosis, edema, glial nodes, perivascular cuffs, and astrocytosis in mice 4–8 weeks old with 100% mortality in 2-day-old mice due to severe inflammation and necrosis of muscle, cartilage and bone marrows prior to neuroinvasion (Aguilar et al., 2011; Phillips et al., 2016; Gardner et al., 2022; Phelps et al., 2017). Suppression of WEEV replication in CD-1 mice using immunotherapy enabled the survival of mice and analysis of neurological sequelae (Bantle et al., 2019). These mice displayed Parkinson's-like symptoms and pathology seen in humans, including a loss of dopaminergic neurons, increased protein aggregation, and persistent neuroinflammatory responses (Bantle et al., 2019). In this model, WEEV replication was confirmed in the olfactory bulb, the cortex, hippocampus, and basal midbrain by day 4 post-infection with prolonged inflammatory response and glia and astrocyte activation 2 months post-infection. A variety of new literature has come out regarding WEEV pathology, especially surrounding its similarities to Parkinson's disease and the chronic consequences of neuroinvasion. Microgliosis, astrogliosis, dopaminergic neuron loss, and a-synuclein protein aggregation are observed across the brain with particular accumulation in the hippocampus and cortex (Bantle et al., 2021).
Despite vast advances in our understanding of disease induced by EEV family members, there have been no studies that have been able to correlate transcriptomic and histopathological features with specific changes in physical, emotional, and behavioral changes seen in humans. Collectively, given the vast similarities indicated in the acute phase of infection, it's likely that therapies could enhance protection against EEV fatality or neurological sequelae.
2.2 TBIThe National Institute of Neurological Disorders and Stroke classifies TBI as an external mechanical force that causes damage to the brain, potentially leading to temporary or permanent disability (NINDS, 2024). The most recent data suggests that over 200,000 TBI related hospitalizations occurred in 2020 and almost 70,000 TBI related deaths occurred in 2021 (CDC, 2024). TBI injury severity varies greatly both across human patients requiring varied animal models to replicate the disease processes and outcomes seen in people. TBI severity can be stratified by the Glasgow Coma Scale (GCS) into mild, moderate, and severe injuries (Vella et al., 2017; Jain and Iverson, 2023). Much of the human research conducted focuses on sports related injuries or combat related injuries (Fehily and Fitzgerald, 2017; Kim et al., 2023; Elder and Cristian, 2009). Even so, the majority of TBIs are caused by falls and frequently impact elderly individuals and young children (Faul et al., 2010). Further, males are over three times more likely to sustain a TBI than females, likely due to more prevalent high-risk behaviors and higher-risk jobs as this sex difference is only present in adults, not children (Alexis et al., 2022). The TBI injury cascade can be generally split into two categories: primary and secondary injury (Davis, 2000). The primary injury reflects the initial mechanical insult, such as axonal shearing, contusion, laceration, and skull fracture. The secondary injury includes the physiological aftermath that causes continued cell damage and death in the hours, days, and years following the initial impact. This includes BBB disruption, hypotension, hyperglycemia, hypoglycemia, changes in intracranial pressure, cerebral edema, peripheral immune cell infiltration, gliosis, and release of excitatory neurotransmitters. These mechanisms lead to the symptoms and outcomes seen in patients and replicated in animal models (Table 3). To replicate human TBI cases, animal models of TBI are adapted to study various injury types and severities (Xiong et al., 2013).

Table 3. Comparison of human TBI cases and laboratory animal models of TBI.
Animal models utilized for TBI research include rats, mice, pigs, rabbits, dogs, swine, sheep, ferrets, monkeys, and cats, which have been evaluated and critiqued by others (Xiong et al., 2013; Cernak, 2005; Ma et al., 2019). Non-human primate models of TBI are essential for accurate modeling of TBI impact on neural damage as these models most closely resemble the human brain; however, these models are limited (Barbay et al., 2021). The most common model of TBI is the rodent (mouse and rat), which we will focus on in this review. Additionally, it is essential to consider that TBI is a heterogeneous injury and that there are many injury models that replicate various pathologies, neurological manifestations, and severity levels. While the classification of TBI is under current refinement including the use of endophenotypes, we aim to review several known severity categories in humans (mild, moderate/severe) and compare human disease pathology to rodent models.
2.2.1 Mild TBIMild TBI is considered a score of 13–15 on the GCS. Mild TBI is the most common type of TBI, with 70%−90% of treated brain injuries falling into the mild category (Cassidy et al., 2004). Furthermore, it is estimated that the incidence of treated mild TBI is 100–300/100,000, however, the true incidence is likely much higher since many mild TBIs are not treated at a hospital (Cassidy et al., 2004). Risk factors for mild TBI include intoxication, low education, age, intimate partner violence, military deployment, contact sports, and socioeconomic status (Alexis et al., 2022; Nordstrom et al., 2013; Gardner and Yaffe, 2015). Frequently, mild TBIs show no abnormalities on CT or MRI scans, yet many patients suffer from a plethora of symptoms. Common symptoms of a mild TBI include post traumatic amnesia, loss of mental alertness, anterograde amnesia, confusion, speech and gait abnormalities, personality changes, a lack of energy, and sometimes a loss of consciousness. Various rodent models of mild TBI, including controlled cortical impact (CCI), weight drop, closed head injury, and fluid percussion injury (FPI) models display many of the outcomes seen in humans (Table 3) (Bodnar et al., 2019). These symptoms included diminished cognitive and motor capabilities, including deficits in spatial learning and memory, increased anxiety-like behavior, and risk-taking behavior. Further, many of the pathological changes in human cases were replicated in mouse models. Pathological changes include accumulation of phosphorylated tau, white matter structure abnormalities, diffuse axonal injury, inflammation, and BBB disruption (Xu et al., 2021; Wu et al., 2020).
2.2.2 Moderate and severe TBIModerate and severe TBIs are classified by having GCS scores of 9–12 and 3–8, respectively. Falls are a leading cause of moderate and severe TBI, particularly for older adults (Iaccarino et al., 2018). Motor vehicle accidents, assaults, and firearm-related injuries also account for many moderate and severe TBIs (Iaccarino et al., 2018; Miller et al., 2020). Loss of consciousness is typically longer with moderate and severe TBI and can last between 30 min and 6 h for moderate TBI and even over 6 h for severe TBI. Common symptomology includes general cognitive impairment, dizziness, confusion, seeing stars, no memory of the injury, persistent or worsening headache, vomiting, nausea, seizures, dilations of pupils, fluid draining from the nose or ears, and/or inability to wake from sleep. Pathological findings can include subarachnoid hemorrhage, subdural hematoma, extradural hematoma, intraventricular hemorrhage, effacement of ventricles, cerebral edema, bradycardia, elevated blood pressure and intracranial pressure, midline shift, cerebral contusion, skull fracture, cerebellar damage, BBB disruption, hippocampal volume loss, increased serum cytokine levels, elevated Glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), and neurofilament light (NfL) deposition. Long-term neurological and psychiatric consequences can include general cognitive impairment, schizophrenia, depression, hallucinations, anxiety, substance abuse, somatoform disorder, adjustment disorder, affective disorder, and general psychiatric diagnosis. Numerous rodent models of moderate and severe TBI have recapitulated human TBI pathology and behavioral deficits (Table 3) (Yu et al., 2009; Pischiutta et al., 2018; Mao et al., 2020).
2.3 NAEarly descriptions of human exposure to NAs occurred through laboratory exposure through inhalation of tabun (ethyl N,N-dimethylphosphoramidocyanidate) (Lopez-Munoz et al., 2008). Exposure to sarin (isopropyl methylphosphonofluoridate), soman (pinacolyl methylphosphonofluoridate), and VX are the most well documented in both patients and animal models (Moshiri et al., 2012). Unlike the aforementioned viruses or TBI models, the mechanism of action and the manifestation of disease is nearly identical across different NAs. The primary mechanism of action of all NAs is the inhibition of acetylcholinesterase activity, leading to accumulation of acetylcholine at neuronal synapses. The excess acetylcholine, termed cholinergic crisis, causes prolonged activation of nicotinic and muscarinic receptor activity, which induces neurotoxic symptoms including muscle cramping, paralysis, headaches, and more (Lopez-Munoz et al., 2008). The accumulation of acetylcholine at the synapses in both the peripheral nervous system (PNS) and CNS leads to widespread CNS damage, with damage in the limbic system, particularly the amygdala and hippocampus, being most noted in the literature (Prager et al., 2014; Shih et al., 2003; Miller et al., 2015). The dentate gyrus of the hippocampus is built of tightly packed cholinergic neurons, therefore, they are highly sensitive to acetylcholine accumulation. Communication between the amygdala and the hippocampus is essential for memory and anxiety behaviors, and these cholinergic neurons play a large role in signal transmission between these portions of the brain (Song, 2023); therefore, it is unsurprising that these regions are significantly impacted by nerve agents (Aroniadou-Anderjaska et al., 2009). While the action of all NAs is similar, the primary differences in disease manifestation are related to the concentration and duration of exposure (CDC, 2017). Unfortunately, high or prolonged low-dosage exposure is highly fatal without immediate medical intervention to mediate seizure damage. The G-series agents (soman and sarin mostly discussed here) are water-like in consistency and easily form vapors, while the V-series agents (VX primarily discussed here) which are a thicker consistency and typically do not vaporize, leading to prolonged presence in the environment (Fan et al., 2024). While not discussed here, it's worth noting that several research articles have used surrogates, which are nonvolatile chemical compounds that inhibit acetylcholinesterase activity but are not as toxic or as strictly controlled as NAs (Finnegan et al., 2021). These compounds are valuable for enabling more laboratory research for therapeutics and biomarker analysis, but are not as toxic as G and V series nerve agents, so they are not the focus of this review.
In the following sections, we will review the animal models of NA exposure, which consist of NHP, rat, and mouse models. Animal models have been reviewed previously (Figueiredo et al., 2018; Pereira et al., 2014). As briefly discussed in future sections, an NHP model is ideal for nerve agent research because humans and primates have conserved nicotinic acetylcholine receptors (Kendrick et al., 2021). Cynomolgus, African green, and rhesus monkeys, as well as common marmosets have been investigated (Despain et al., 2007). Some models that have not been as well evaluated, such as the baboon model, have shown significant airway decline and neuromuscular junction activity, but the effects were inconsistent due to anesthetization with phenobarbital which inhibits some of the clinical presenting signs of NA toxicity, including muscle fasciculation and seizures (Anzueto et al., 1990). Collectively, it has been suggested that NHPs are the best model for NA intoxication due to similar levels of organophosphorus metabolizing carboxylesterases to humans though new humanized mouse models are being developed to better mimic human responses and reduce some of the ethical challenges of NHP experimentation (Marrero-Rosado et al., 2021; Tressler et al., 2024).
2.3.1 G-series nerve agents (GA, GB, GD)Several instances of G-series NA exposure have been documented in humans, but the extent of exposure and resulting neurological sequelae are not well characterized. Notable use occurred in Northern Iraq (Iraq, Iran War), Syria, and Damascus (Balali-Mood and Saber, 2012; Rosman et al., 2014; Thiermann et al., 1999). Documentation of sarin exposure in humans has been primarily on accidental exposures (Duffy et al., 1979) though several long-term studies of sarin exposure have been conducted on victims of the Tokyo and Matsumoto subway terrorist attacks (Duffy et al., 1979; Okumura et al., 1996). Inhalation and ingestion are the most toxic routes of NA exposure and induce faster, more severe symptoms than dermal contact which can take hours for symptoms to onset (Vucinic et al., 2017). Sarin, tabun, and soman are highly volatile and lethal doses are estimated to be between 10–500 mg-min/m3 (Table 4). Acute symptoms of exposure include headache, nausea, vomiting, and seizures (CDC, 2018). Prolonged symptoms of G-series exposure include depression, anxiety, mood swings, memory impairment, and cognitive and behavioral changes (Figueiredo et al., 2018; Levin and Rodnitzky, 1976; Rosenstock et al., 1991; Wesseling et al., 2002; Savage et al., 1988; Roldan-Tapia et al., 2005). Some evidence has suggested that Gulf War illness, characterized by broadly defined features such as tiredness, pain, memory impairment and imbalance in >250,000 warfighters, may be partially due to low levels of sarin exposure (Haley et al., 2022; Elhaj and Reynolds, 2023). These long-term effects are currently hypothesized to be due to intracellular cytotoxicity that correlates with the white matter edema observations in human cases following the Tokyo sarin attacks, but post mortem analysis of NA-exposed humans is limited (Bhagat et al., 2001, 2005). Collectively, early detection and antiseizure administration are the most crucial mediator of severe OP neurotoxicity (Gupta, 2020).
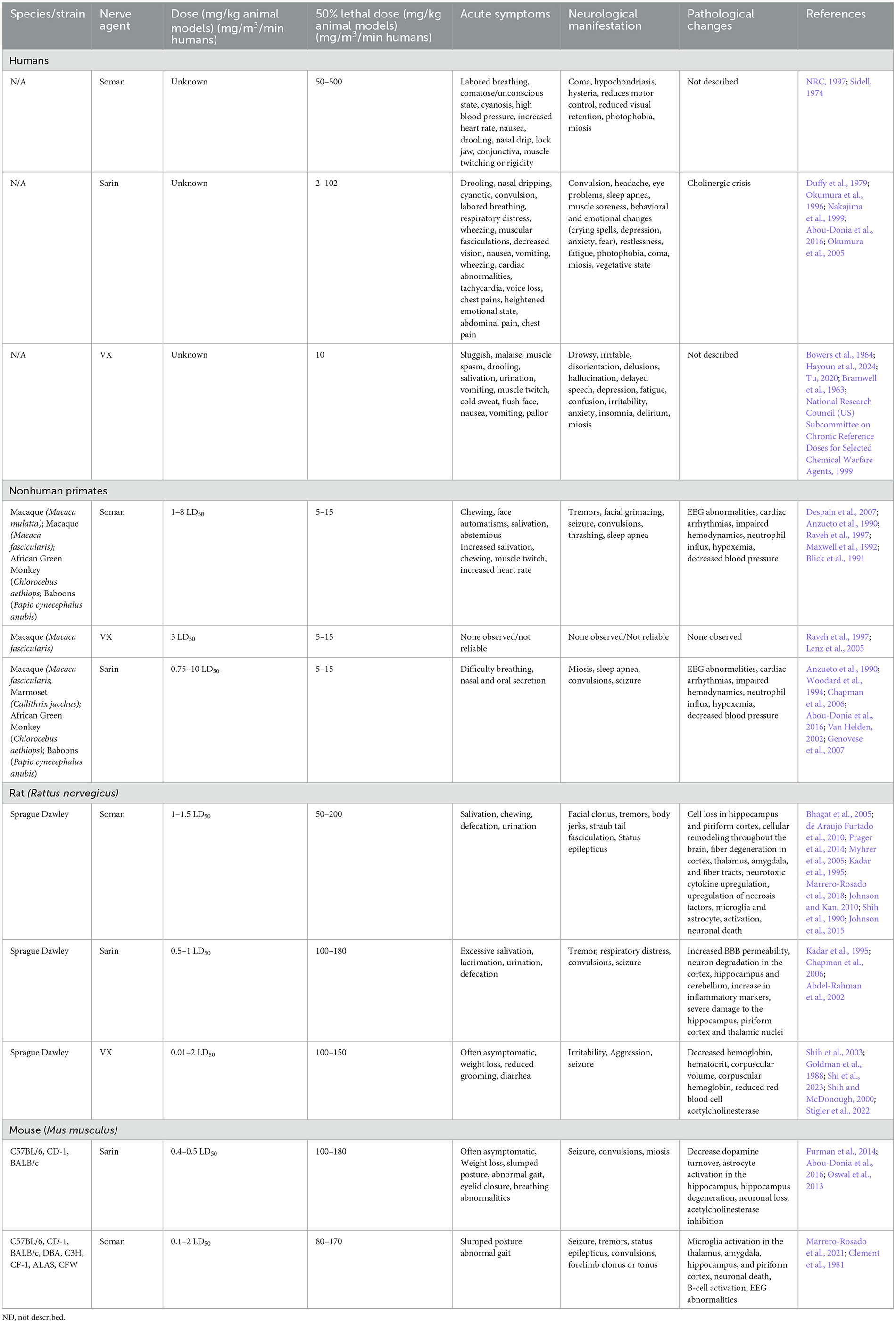
Table 4. Comparison of human cases and laboratory animal models of NA.
Soman exposure in NHPs has been well-established in several models (rhesus, cynomolgus, baboons, African green) to induce tremors, seizures, and muscle spasms (Despain et al., 2007; Raveh et al., 1997; Woodard et al., 1994; Maxwell et al., 1992). Immediate symptoms include chewing, increased salivation, and facial spasms with symptoms progressing to limb twitching, persistent tremors, convulsion, seizure, thrashing, and slumped posture (Despain et al., 2007). Without medical countermeasures to mediate seizure and acute toxicity, NA exposure is typically lethal. The lethal dose reported in African green monkeys was ~7.15 μg/kg, which is consistent with ranges from 5–15 μg/kg in rhesus, cynomolgus, and baboons (Table 4). Seizure activity is closely correlated with administered dosage of soman, where lower dosages result in less severe seizures and other neuromuscular changes, while higher dosage animals are prone to more frequent and longer lasting seizures, apnea, and cyanosis (Despain et al., 2007). Apnea, cardiac arrhythmia, and decreased blood pressure following soman exposure have been most well characterized in NHPs (Despain et al., 2007; Raveh et al., 1997; Woodard et al., 1994; Maxwell et al., 1992). The prolonged activation of acetylcholine receptors in humans and NHPs induces fatal neurotoxicity and, ultimately, death.
Symptoms of nerve agent poisoning in rodents resemble that of humans and NHPs with respiratory challenges, involuntary secretions, chewing, salivation, diarrhea, muscle fasciculation, tremors, convulsions, and seizures (Table 4). Soman can cross the BBB rapidly leading to increases in acetylcholine followed later by glutamate and subsequent neurotoxicity (Bhagat et al., 2005; Shih and McDonough, 1997). A single high dose of sarin or soman induces neuronal loss in rats dependent on seizure activity (McLeod et al., 1984; Petras, 1994). Pathological signs of injury occur soon after injury (4 h) but appear to worsen over time with notably increased severity at 3+ months post injury in the hippocampus, piriform cortex, and thalamus (Kadar et al., 1995). Single doses of sarin induce edema widespread \throughout the brain (Testylier et al., 1999). Extensive pathology of soman-exposed rats with memory deficits showed significant loss of neurons and interneurons and an increase in activated astrocytes and microglia in the hippocampus 90 days post-exposure (Reddy et al., 2020; Marrero-Rosado et al., 2018). BBB disruption and permeability significantly increase with exposure to NA across the brain; however, these impacts appear to be acute while the dysregulation of acetylcholine and muscarinic receptors appears to persist beyond 90 days post-injury. Persistent BBB disruption following NA exposure has not been well documented. Currently, BBB disruption appears to be at its height in the acute phase post-exposure and may be associated with convulsions and seizure typically seen acutely following exposure (Gupta, 2020). However, BBB restoration following injury in general can take time and depends on several factors, including severity of disruption and neuroinflammation. NA exposure is likely not an exception to this. In addition to pathological markers, there has been significant evaluation of neurotoxicity in rodent brains at various timepoints post exposure that display increased cytokine production, oxidative stress, and locomotor activity which becomes more apparent with increasing concentration of nerve agent (Henderson et al., 2002; Abu-Qare and Abou-Donia, 2002; Nieminen et al., 1990; Johnson and Kan, 2010). A review of animal models exposed to soman highlighted that in both rat, guinea pig, and nonhuman primate NA exposure models, only animals with seizures developed neuropathology and these seizures are the best variable for predicting neuron loss (Abdollahi and Karami-Mohajeri, 2012; Jett et al., 2020).
Unique challenges of NA exposure model development are the differing baseline levels of cholinesterase activity between inbred and outbred rodent strains. It has been relatively well established that C57BL/6 mice and C3H/He have less acetylcholinesterase activity in comparison to DBA/2 and BALB/c mice (Atalayer and Rowland, 2010). Genetic differences, including cholinesterase levels, appear to contribute to NA toxicity, especially regarding lethal dose and percent mortality (Matson et al., 2018; Furman et al., 2014). An additional challenge is the presence of serum carboxylesterase activity in rodent models of NAs. These compounds aid in degradation of NAs, thereby reducing the total NA concentration, but this activity appears to be absent in human cases. Differences in rodent response to NAs could also be sex dependent, as neurodegeneration and gliosis 4 months post exposure to soman is more significant in female animals than male animals (Gage et al., 2021).
2.3.2 V-series nerve agents (VE, VG, VM, VR, VX)The largest evidence of VX exposure consists of nearly 100 warfighters presenting ill-defined symptoms, including “altered awareness,” reduced intellectual ability, slowed movements, anxiety, and confusion (Bowers et al., 1964). V- series NAs induce a variety of symptoms including seizure, salivation, urination, vomiting, spasms, muscle twitching, and tremors (Hayoun et al., 2024). Incidence of seizure is lower in both humans and animals exposed to VX or VR compared to the G-series agents (Shih et al., 2003; Fawcett et al., 2009). Similar to the other OPs, chronic evaluation of nerve agents in humans is limited, but amnesia and behavioral changes may persist years after exposure (Nozaki et al., 1995). While human cell culture models of NAs are limited, a unique study performed a microarray analysis of human neurons and astrocytes exposed to VX and soman, respectively, and identified significant upregulation of apoptosis and inflammatory cascades independent of in vivo hallmarks of seizures and acetylcholine dysregulation (Hoard-Fruchey et al., 2020). Gene expression changes of astrocytes were not dependent on the agent used, but neurons appear to be significantly separated with both datasets showing significant upregulation of genes associated with the inflammatory response. It should be noted though that cell death was not induced by VX or soman in either cell population.
Multiple models for VX exposure exist including goats, guinea pigs, swine and mice. Observations in these animals are consistent with dose-dependent impacts of G-series agents, where facial twitches, chewing, tremors, seizures, but appear to be brief and all animals typically survive (Fawcett et al., 2009; Langston and Myers, 2016; O'Donnell et al., 2011). Ultimately, NHP and guinea pig models, previously reviewed (Pereira et al., 2014), appear to be the most reliable over rat and mouse models. Of the rodent models of VX exposure, there were minor changes in acute toxicity, histopathology, and weight, leading to authors conclusion of unreliable results with subcutaneous administration of sublethal doses (Goldman et al., 1988; Atchison et al., 2001). Minimal exploration of chronic aspects of VX exposure have been explored.
3 Comparative neuropathologyIndicated throughout this review, there are overarching similarities across the three neuropathologies in terms of symptoms and pathology. In humans, the symptomatic manifestations of disease often include seizure, confusion, behavioral or emotional changes. In following sections, we will compare disease progression of EEVs, TBI, and NA.
3.1 Overlapping clinical sequelae in humansDisease manifestations of EEVs, TBI, and NA include overlapping symptoms and neuropathologies, which highlight the potential for both research gaps in neurological disease mechanisms as well as therapeutic potentials. These illnesses induce mild symptoms such as fever, confusion, headache, shock, neck pai
留言 (0)