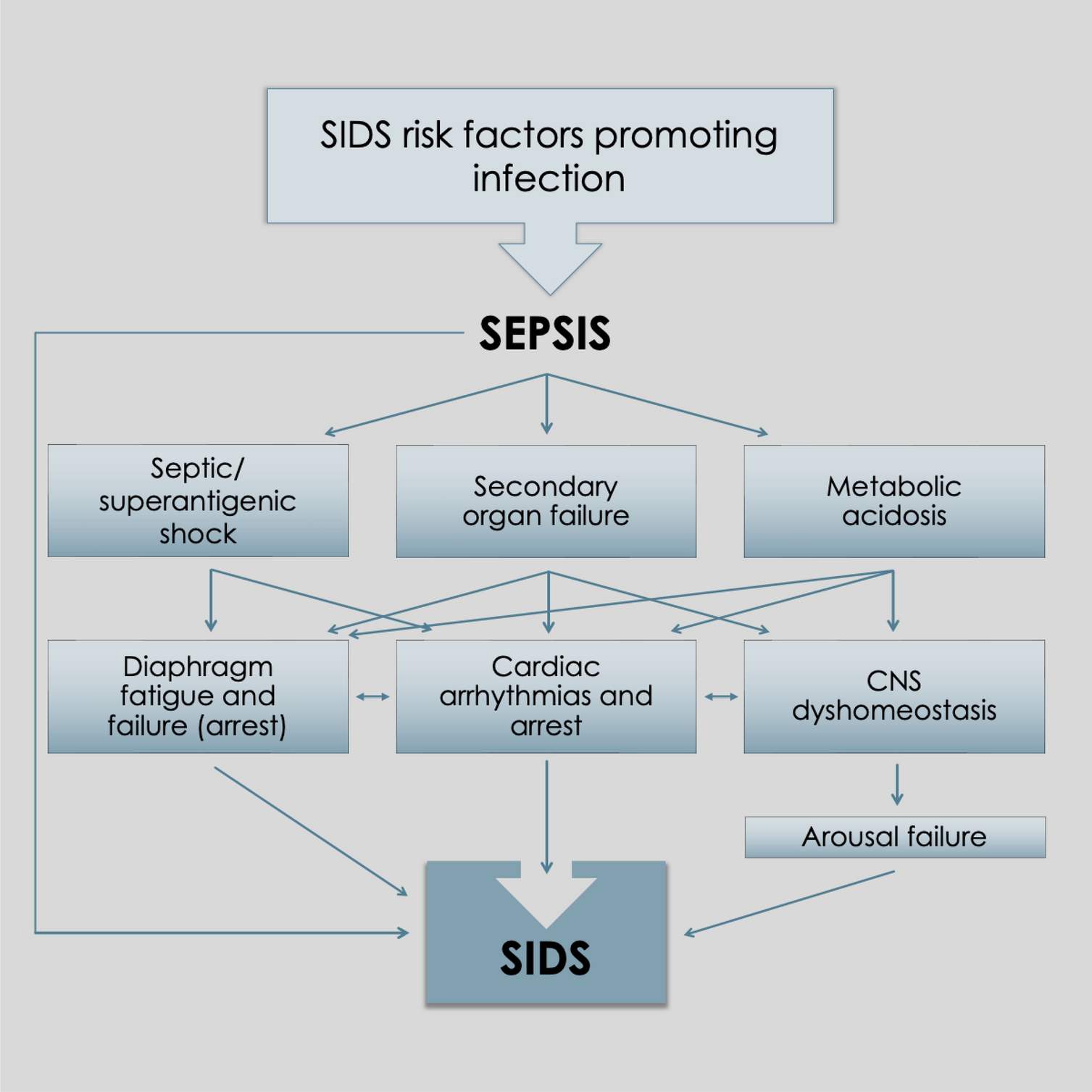
Sepsis, diaphragmatic failure and lethal arrhythmias
Sepsis causes end organ damage through a multitude of biochemical processes, and skeletal muscles such as the limbs and diaphragm feature unexceptionally. Both become atrophic and weakened, resulting in reduced contractility and endurance [51, 52]. This is important because the diaphragm is the primary muscle of respiration. Diaphragm fatigue (or diaphragm dysfunction, DD), distinguished from weakness by its reversibility with rest, is common among intensive care unit (ICU) patients and can occur rapidly and profoundly. Acute bacterial infections can diminish diaphragm contractile strength by as much as 80% within 24 h of ICU admission [53]. Severe sepsis is often complicated by concomitant hypoxemia, hypovolemia, acidosis, and comorbid disease [54]. Evidence supporting peripheral, or type II, hypercapnic (ventilatory pump) failure in sepsis was first demonstrated in anesthetized dogs by Hussain et al. in 1985 [55]. As measured by transdiaphragmatic pressures, airflow rates and diaphragmatic electromyography, all animals given intravenous Escherichia coli endotoxin developed rapid respiratory insufficiency followed by tachypnea, bradypnea, and then sudden apnea leading to cardiac arrest within 1‒2 min. Death took only 150‒270 min. Hussain went on to declare that, in septic shock patients [ones not receiving mechanical ventilation], peripheral respiratory failure was the most important cause of death, even after hemodynamic aberrations had been corrected [56]. Sepsis-associated acute lung injury further contributed to the fatigue and failure by increasing ventilatory workloads.
Although the pathological mechanisms responsible for diaphragm weakness, fatigue, and failure in sepsis are complex and beyond the scope of this paper, they generally include activation of the proteolytic pathways as well as overproduction of inflammatory cytokines, prostaglandins, reactive oxygen species and nitric oxide. At the cellular level, this leads to structural myofiber injury (inducing organ atrophy and myopathy), impaired action potential generation and propagation through failure of neuromuscular and intradiaphragmatic depolarization, and disrupted excitation‒contraction coupling or damage to the contractile machinery itself. At the molecular level, contractile dysfunction occurs through altered Ca2+ homeostasis, which is influenced by endotoxins, acidosis, electrolyte disorders, and muscle fatigue itself, among other factors [57,58,59]. In experimental endotoxemic rats, reduced diaphragmatic contractility and endurance were associated with a decreased resting membrane potential and prolonged muscle relaxation time [54]. Although subsequent diaphragm neuromuscular inexcitability contributes to further fatigue [60], pathological excitation also occurs. Other than persistent hiccups, however, there is a paucity of data. A few case reports did reveal a variety of “hyperexcitation disorders”, including diaphragm spasms, tics, tremors, and myoclonus, as well as low- and high-frequency diaphragmatic flutter [61, 62]. Patients of all ages are affected by these apparently obscure disorders and generally present with chest or epigastric pain, sometimes with visible epigastric pulsations, and a variety of gastrointestinal and respiratory symptoms. The latter ranges from dyspnea and transient apneas in tetraplegic patients with diaphragm spasms [63], to respiratory distress and prolonged apneas requiring ventilatory support just hours after birth in neonates with respiratory flutter [62]. In addition, in a compelling pediatric case recently published, a sustained diaphragm spasm (akin to a tetanic cramp-contracture) was proposed to cause sudden unexpected respiratory arrests in children with severe diaphragm fatigue [64].
Like the heart, the diaphragm is a vital pump essential to life. Suddenly impaired cardiac output (cardiac arrest) from asystole or malignant cardiac arrhythmias is known to cause sudden unexpected deaths (for example, from ventricular fibrillation or various unstable ventricular tachycardias). Similarly, extremely rapid or ineffective diaphragm contractions which significantly impair lung alveolar ventilation could also lead to sudden deaths by respiratory failure. Sepsis, which is known to contribute to cardiac arrhythmias and arrests [65], is also suspected to impair diaphragmatic function through an analogous mechanism contributing to critical diaphragmatic failure in sudden infant deaths (i.e., unstable diaphragm arrhythmias) [64, 66]. In fact, DD (and work overload) is induced and exacerbated by several SIDS risk factors, including viral respiratory or gastrointestinal infections [diaphragm damage from myositis and myopathy (vide infra) with concomitant dehydration, acidosis, and electrolyte disorders], the young infancy period (harder working underdeveloped, untrained ventilatory muscles), rebreathing exhaled gases (causing net hypoxemia, hypercapnia, and respiratory acidosis; all worsening diaphragm contractility), tobacco smoke exposure, prone positioning, and rapid eye movement (REM) sleep (CNS inhibition of airway dilator and accessory respiratory muscles) [66].
It is important to point out that diaphragm contractile dysfunction from hypoxemia and hypercapnia creates positive feedback cycles exacerbating both conditions. This is because of fatigue-induced alveolar hypoventilation. In other words, diaphragm pump insufficiency caused by hypoxemia begets a further drop in blood oxygen levels. Ultimately, escalation of this unstable process could culminate in rapidly critical hypoxemia, causing sudden respiratory and cardiac arrests similar to the Hussain dog experiments [55]. This is also consistent with the features of many pediatric deaths in general, including unwitnessed ones like SIDS, given many are unexpected, sudden in onset, and rapid. Death by respiratory arrest would be silent too, another feature of SIDS.
Importantly, nicotine has a potent direct effect on skeletal muscles. In excess, even with minute ingestions in young children, death occurs rapidly, via rapid ventilatory muscle paralysis (again, by tetanic-like diaphragm arrest) [67]. In spontaneously breathing, anesthetized dogs oral and intravenous nicotine, peripheral respiratory arrests occurred in less than 15 min, causing death if not artificially ventilated [68]. Furthermore, ex vivo rabbit, frog, and cat limb muscles exposed to nicotine developed tetanic contractures [69,70,71]. Given that the diaphragm is also a skeletal muscle and high nicotine levels have been found in the tissues of SIDS victims [72], it is important to consider the potential for sudden diaphragm arrest (by contracture) to occur in infants exposed to tobacco smoke, especially in the presence of other diaphragm fatiguing DD factors. Nicotine could effectively lower the cramp threshold.
Evidence for of diaphragm myopathy in SIDS is provided by several histological studies [73, 76]. Focal, segmental, and diffuse diaphragm myofiber disruptions and contraction band necrosis, along with fibrotic scars in some near-miss SIDS cases were first reported by Kariks in 1989 [73]. Inflammatory cell infiltration (myositis) was not present in this study, suggesting a rapid, terminal onset of terminal changes. Contraction band necrosis (CBN), indicative of terminal asphyxia and anoxia, was confirmed in 82% of 242 SIDS cases in the this systematic study and later was corroborated by the three others. Importantly, despite the peculiar findings of “extreme compaction of sarcomeres in hypercontracted segments” in CBN, a mechanism has never been proposed. Research in this area inexplicably appears to have stalled. It also remains undetermined whether these myopathic changes might exist in sepsis and nicotine deaths. Regardless, given such compelling findings, it is reasonable to mandate diaphragm histology in all cases of sudden pediatric death, especially those with respiratory and / or gastrointestinal viral infections, bacterial infections, sepsis or nicotine exposure.
Further evidence of diaphragmatic histopathological changes during infection was provided in four other reports. The first, by Eisenhut (2011), documented focal infiltrates, myofiber destruction, and myocyte necrosis, and regeneration and in a 5-month-old with respiratory syncytial virus (RSV) infection, who had died in the hospital from sudden and unexpected respiratory arrest [76]. This was nearly identical to another paroxysmal respiratory arrest reported recently in an infant with RSV bronchiolitis [77]. Autopsy findings could not explain the death; however, diaphragm histology was omitted (not unusual because autopsy guidelines did not mandate this). A third report revealed, that three young children, aged 3 days to 5 years, who died unexpectedly from respiratory arrest, had exhibited a combination of diaphragm necrosis, inflammatory infiltration, and fiber regeneration at autopsy [78]. The 5-year-old previously healthy girl, who complained of chest and abdominal pain just before collapsing, had a hemoglobin level of 10.7 g/dL and a pH of 6.59. In addition to this extreme acidosis, the anemia could have contributed to diaphragm fatigue and failure because of the reduced blood oxygen-carrying capacity of blood. The fourth paper, a case‒control study examining the association of severe COVID-19 infection with the respiratory muscles of critically ill adult intensive care unit (ICU) patients, revealed direct viral infiltration of the diaphragm with fibrosis and regeneration in the case patients only [79].
Taken together, the above findings suggest that viral myositis and the myopathic changes in the diaphragms of young children with respiratory infections could have interfered with excitation–contraction coupling or electromechanical function of this organ, leading to escalating diaphragm fatigue and terminating in death by paroxysmal diaphragmatic failure. Because of the suddenness, it is possible such respiratory arrests may have occurred by critical hypoxemia-induced pathological diaphragm hyperexcitation, in the form of a rapid onset sustained cramp culminating in death by sudden respiratory arrest. This would explain the 5-year-old’s chest and abdominal pain prior to arresting. This could be the source of the hypercontraction injury and contraction band necrosis commonly seen in SIDS and would be consistent with the sustained contractions of putative diaphragm cramp-contracture.
By comparison, “myocardial electrical instability,” in the form of cardiac arrhythmias, was discussed in a 2019 summary report on viral myocarditis [80]. Sudden cardiac death, which is more common in males under 40 years of age, is particularly concerning because the incidence of occult myocarditis at autopsy was as high as 44%. Moreover, infectious myositis of the myocardium in sudden unexpected deaths is more common than thought with the same occurring in the diaphragm leading to the same fatal outcome.
Metabolic acidosis, diaphragmatic failure and lethal arrhythmias
Sepsis disrupts acid‒base balance via a variety of mechanisms leading to metabolic acidosis. This fatigues skeletal muscles of the limbs, trunk and diaphragm, which is particularly aggravated by hypoxemia and hypercapnia [81, 82]. In critically ill patients, metabolic acidosis generally occurs through the accumulation of acidic anaerobic metabolites, which are increased by cytokines and other inflammatory mediators during infection, as well as bicarbonate loss in severe diarrhea or renal insufficiency. It is often accompanied by electrolyte imbalances resulting from fluid loss and disrupted renal function. Tissue hypoperfusion with reduced lactate clearance by the liver and kidneys leads to lactic acidosis, the primary cause of inpatient metabolic acidosis. Elevated lactate levels serve as a prognostic marker of disease severity and mortality. Although the causal relationship is unknown, decompensation of comorbid conditions, vascular smooth muscle dysfunction, myocardial depression and cardiac arrhythmias are most often cited [83]. Less is known about respiratory failure because it is often concealed, as these severely ill patients are already receiving lifesaving mechanical ventilation.
Extracellular pH has a major influence on skeletal (and heart muscle) electrophysiology, similar to the mechanisms already described in sepsis, leading to pathological inexcitability (atrophy and fatigue) and hyperexcitation. As alluded to above, ventricular fibrillation (VFib) can be considered a form of muscle cramp. Its quivering, arrhythmic and ineffective contractions significantly impair cardiac output, causing pump failure. This malignant arrhythmia is sensitive to the acid‒base balance of blood. In anesthetized dogs, infusion of organic acids from a physiologic pH of 7.42 to 7.21 progressively lowered the threshold for VFib [84] (which was reversed by alkaline infusion) [84]. This could have occurred through an altered conductance of voltage-gated ion channels, for example, the hERG1 potassium channel (expressed in the heart) [85] or the SCN5A sodium channel (heart and skeletal muscles) [86]. The latter plays a critical role in physiological excitation but is extremely sensitive to low pH [87]. Both channelopathies have been suspected to cause sudden unexpected deaths by inducing cardiac arrhythmias, primarily long QT syndrome. Similarly, unstable diaphragmatic arrhythmias may also be triggered. Unfortunately, however, this organ has been entirely omitted from ion channel tissue distribution studies. This might explain why only 2% of 93 SIDS victims had a SCN5A channel defect in their myocardia (and not any higher) [88]. In other words, a higher prevalence might have existed had their diaphragms been examined which illustrates restricted thinking in mainstream research.
Metabolic acidosis in young children with inborn errors of metabolism has also been reported in association with sudden deaths (or near-deaths). Some of these disorders involve defects of mitochondrial electron transport proteins causing cardiomyopathy and skeletal muscle myopathy (e.g., myalgia, hypotonia and fatigue) [89, 90]. In these case reports, sudden respiratory distress, including apnea and labored, agonal breathing, occurred prior to cardiopulmonary arrests. Interestingly, RSV and rotavirus infections were noted in addition to severe acidosis, some with copious vomiting and diarrhea. Given this and the associated myopathies presumably involving the diaphragm, peripheral respiratory failure could have been responsible for the deaths/near-deaths.
Pediatric deaths from severe diarrheal illness in developing countries, which also occur rapidly and unexpectedly, are often associated with bicarbonate-loss -induced hyperchloremic acidosis (as well as hyponatremia and hypokalemia) [91, 92]. Among patients seen in hospitals, terminal pathological mechanisms include VFib and rapidly progressive respiratory distress; however, there is a paucity of information on the latter other than “complications of respiratory muscle fatigue” (again, obscured by mechanical ventilation). With metabolic acidosis, the fatigue manifests in response to CNS-mediated Kussmaul’s respirations: compensatory tachypnea and hyperpneas to “blow off” CO2, that can deteriorate in extremis to agonal breathing. Similarly, this occurs in diabetic ketoacidosis, where many deaths are also sudden and unexpected, even in the hospital setting [93]. pH is extremely low and associated with a hyperosmolar state featuring extremely elevated lactate levels, hypokalemia, hypomagnesemia, and hypophosphatemia. In a retrospective observational case study involving patients of all ages, including infants, at least 30 of 69 died by witnessed sudden respiratory arrest (terminal apnea). Most, but not all these respiratory arrests, were preceded by mental status changes suggestive of terminal cerebral edema (thought to cause central respiratory arrest). However, in 20 patients, including most infants and toddlers, there was no change before the respiratory arrest. This suggests the deaths may not have been centrally induced and is consistent with other authors’ conclusions [94]. Given that increased work of breathing occurs in severe acidosis and that the resulting diaphragm fatigue is exacerbated by concomitant hypovolemia, hypoxemia, hypercapnia, and electrolyte disorders, it is reasonable to propose that peripheral respiratory failure, caused by sudden diaphragmatic arrest, could be a terminal pathological mechanism in severe acidosis.
The mechanisms leading to skeletal muscle dysfunction in metabolic (and respiratory) acidosis are quite complex. Essentially, intracellular acidosis increases ionized calcium bound within sarcoplasmic reticulum (SR) stores and reduces its SR uptake. The lack of available SR calcium disrupts excitation‒contraction coupling, leading to muscle fatigue by both a reduction in contractile force and prolongation of the muscle relaxation phase, a process that is load sensitive (i.e., the heavier the workload is, the less muscle shortening and the longer the relaxation phase) [95,96,97]. This is known as negative lusitropy and is mirrored by cardiac diastolic dysfunction. Under higher heart rates, the dysfunction is exacerbated by a delay in left ventricular relaxation, leading to reduced cardiac output. This is manifested by worsening exercise tolerance and congestive heart failure [98] and puts heart failure patients at risk for sudden arrhythmias and cardiac arrest [99].
In terms of the diaphragm, ex vivo studies have revealed that fatigue-induced tetanic contracture develops when the relaxation time is excessively prolonged [59]. This is exacerbated by acidosis [97], endotoxins [54] and fatigue itself. (Notably, DD improves with methylxanthines such as theophylline and caffeine, which have been used for over 50 years to treat apnea and periodic breathing in preterm infants [100].) In vivo, this delay could be problematic under higher-frequency breathing: when relaxation takes longer than does the expiratory phase of the respiratory cycle, incomplete return to the resting position occurs. Consequent air trapping (breath stacking) and hyperinflation would reduce the mechanical advantage at the diaphragm, thereby exacerbating the DD. Furthermore, given that diaphragmatic perfusion occurs primarily during the relaxation phase, higher-frequency breathing could then lead to metabolic mismatch within the organ [96], thereby contributing to further fatigue in another DD positive feedback cycle (in addition to the hypoxemic‒hypercapnic one already discussed). With worsening fatigue by dehydration, acidosis, endotoxins, electrolyte disorders, and myopathy, the relaxation delay could explain pathological excitation. If it occurs as transient diaphragm spasm apneas, the ensuing hypoxemia would be survivable. However, when sustained as a cramp-contracture respiratory arrest, the severe hypoxemia could be fatal (by secondary cardiac arrest).
Importantly, in vitro experiments have shown that lactic acidosis decreases diaphragmatic contractility but only at very low “extraphysiological” pH values of 6.80 [101]. However, McGaffey’s report of an average pH of 6.15 in forty SIDS victims suggests otherwise. Thus, extrapolating in vitro data to in vivo animal or human studies requires special caution (vide infra). Kimmoun et al. found that no survival has been reported for severe lactic acidosis with shock under pH 7.0 [83].
Although human sepsis causes metabolic acidosis and can result in cardiac and diaphragmatic failure, animal experiments in rats have been unsuccessful in demonstrating this adverse outcome, with respiratory acidosis causing diaphragm failure but metabolic acidosis not [102]. As alluded to above, limitations in animal and/or in vitro experimental conditions make extrapolation to the human model imprudent.
Other causes of vital-organ failure in sepsis
The other causes mentioned above, namely cardiac arrhythmias/asystole with increased risk from genetic predisposition, cardiac asystole secondary to sepsis-induced hyperkalemia [103,104,105], respiratory failure (CNS-based) [106] and genetically predisposed mitochondrial energy failure (inborn errors of metabolism) [107], have been reviewed elsewhere and are, therefore, not included in this paper. However, it is noteworthy that while “infection” might be listed as an accompaniment, mention of sepsis is rare, if it is ever made, in articles on inborn errors of metabolism and SIDS.





留言 (0)