General
Oligonucleotide primers and DNA sequencing services were provided by Eurofins Genomics. Restriction enzymes and PrimeSTAR MAX DNA polymerase were purchased from Takara Bio Inc. Solvents and chemicals were purchased from Wako Chemicals, Merck KGaA and Hampton Research, unless noted otherwise. Polymerase chain reaction (PCR) was performed using a TaKaRa PCR Thermal Cycler Dice Gradient system (TaKaRa Bio). NMR spectra of compounds were recorded on Bruker AVANCE III HD 900 MHz and ECX-500 MHz (JEOL) spectrometers.
Construction of plasmids for LmbF and CcbF expression
The lmbF gene was PCR-amplified from the chromosomal DNA of the lincomycin-producing-type strain Streptomyces lincolnensis ATCC 25466, using the following primers: forward CCGCATATGTCCGACTTAGCTGCCGTTGATGC and reverse CCGCTCGAG CCGGTACCGCCACTCGGCCGCGG. The lmbF gene was inserted into the pET42b vector (Novagen) via the NdeI and XhoI restriction sites. The resulting plasmid was used to produce C-terminally His8-tagged LmbF. The ccbF gene was PCR-amplified from chromosomal DNA of the celesticetin-producing-type strain Streptomyces caelestis ATCC 15084, using the following primers: forward CCGCATATG TCCGACTTAGCTGCCGTTGATGC and reverse CCGCTCGAGGCGGGGCTGCCAGGCGCGTGAGG. The ccbF gene was inserted into the pET42b vector (Novagen) via the NdeI and XhoI restriction sites. The resulting plasmid was used to produce C-terminally His8-tagged CcbF.
Expression and purification of CcbF, LmbF and their variants
The pET42b plasmids for the expression of CcbF, LmbF and their variants were transformed into Escherichia coli BLR(DE3) harbouring the pGro7 plasmid. The resulting strains were cultured in LB medium supplemented with 34 mg l−1 chloramphenicol and 50 mg l−1 kanamycin sodium at 37 °C, with shaking at 160 r.p.m. When the optical density at 600 nm (OD600) reached 0.6, the cell cultures were cooled on ice for 30 min, then isopropyl β-d-thiogalactopyranoside (IPTG; 0.3 mM) was added to induce the target protein expression and the cultures were maintained at 16 °C, 160 r.p.m. After 18 h of post-induction incubation, cells were collected by centrifugation at 5,500g for 10 min and suspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM imidazole and 10% glycerol. The cell suspension was sonicated for 5 min on ice. After the cell debris was removed by centrifugation at 20,000g for 30 min, the supernatant was mixed with 1 ml of TALON resin and loaded onto a gravity flow column. Unbound proteins were removed with 100 ml of lysis buffer containing 20 mM imidazole, then the His-tagged protein was eluted with lysis buffer containing 300 mM imidazole. For the in vitro assay, the eluted enzymes were concentrated after buffer exchange to 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM imidazole, 0.5 mM EDTA, 1 mM dithiothreitol (DTT) and 10% glycerol, using a 30-kDa Amicon Ultra-15 filtration unit (Millipore).
For crystallization, the His-tag-purified enzymes were applied to a 6-ml Resource Q anion exchange chromatography column (4 °C, Cytiva) and a HiLoad 16/60 Superdex 200 prepacked gel filtration column (4 °C, GE Healthcare), and eluted with a solution containing 20 mM HEPES (pH 8.0), 100 mM NaCl and 1 mM DTT. The resulting eluate was concentrated to 7 mg ml−1 using an Amicon Ultra-4 (molecular weight cutoff, 30 kDa) filter at 4 °C. The purity of the proteins was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE; Supplementary Fig. 12), and the protein concentrations were determined with a SimpliNano microvolume spectrophotometer. The stability of the variants was tested by incubating the enzymes at 37 °C for 1 h. The aggregated proteins were separated by centrifugation, and the proteins in the supernatants and precipitates were analysed by SDS–PAGE (Supplementary Fig. 13). Notably, the stabilities of the LmbF and CcbF variants were comparable to those of the wild types.
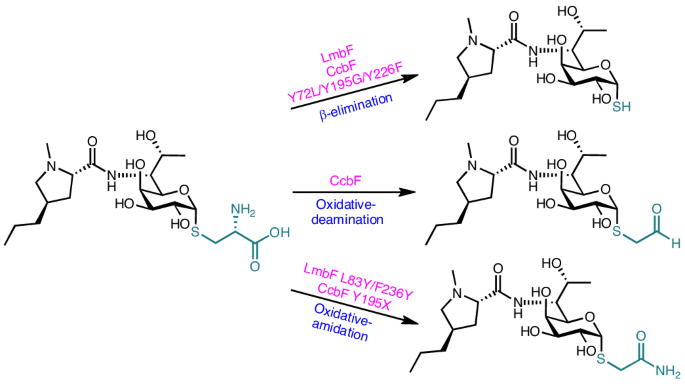
In vitro assays of LmbF, CcbF and their variants
The standard enzymatic reaction of LmbF with substrate 4 was performed in a 12.5-μl volume containing 100 mM potassium phosphate (KPi, pH 8.0), 40 μM substrate 4, 0.5 mM EDTA, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), 0.5 mM PLP and 0.2 μM enzyme, and incubated for 5 min at 37 °C. For the in vitro analysis of the multiple mutants of LmbF, 12.5-μl reaction mixtures containing 100 mM KPi (pH 8.0), 40 μM substrate 4, 0.5 mM EDTA, 0.5 mM DTT, 0.3 mM S-adenosyl-methionine (SAM), 0.1 mM PLP, 4 μM wild type or LmbF variants, and 10 μM of LmbG to convert unstable product 6 to stable lincomycin A (1), were incubated for 60 min at 37 °C. The standard enzymatic reaction of CcbF and its variants with substrate 4 was performed in a 12.5-μl volume containing 100 mM KPi (pH 8.0), 40 μM substrate 4, 0.5 mM EDTA, 0.5 mM DTT, 0.1 mM PLP, 0.1 mM SAM, 20 μM wild type or mutant CcbF, and 20 μM of LmbG to convert the unstable product 6 to stable lincomycin A (1), and incubated overnight at 37 °C. Liquid chromatography mass spectrometry (LC–MS) samples were injected into a Shimadzu Labsolution LCMS 8045 system. A COSMOSIL 5C18-MS-II packed column (2.0 × 50 mm, 5 μm; Nacalai Tesque) was used for separation. Gradient elution was performed with solvent A (1 mM ammonium formate, pH 9.0) and solvent B (CH3CN), with a flow rate of 0.2 ml min−1 (min, % of B: 0 min, 10%; 3 min, 10%; 12 min, 100%; 15 min, 100%), followed by 5 min of equilibration with 10% solvent B before the next analysis.
For the O2 saturation reaction, a premixed solution containing 100 mM KPi, 0.5 mM EDTA, 0.5 mM DTT, 0.3 mM SAM and 0.1 mM PLP was saturated with O2 by bubbling O2 gas for 30 min before reactions, then 20 μM wild type or mutant CcbF was incubated in 25-μl reaction mixtures containing 100 mM KPi, 0.5 mM EDTA, 0.5 mM DTT, 0.3 mM SAM, 0.1 mM PLP, 40 μM substrate 4, 10 μM LmbG, prepared from the O2-saturated solution for 120 min at 37 °C under an O2 atmosphere. The reactions were quenched by adding 100 μl of methanol. Reactions without O2 bubbling were performed as negative controls. The samples were analysed with the same method as that used for the standard reaction.
Labelling experiments
For the 18O2 labelling experiment, 25-μl reactions were performed in 500-μl tubes containing 100 mM KPi (pH 8.0), 40 μM substrate 4, 0.5 mM EDTA, 0.5 mM DTT, 0.5 mM PLP and 0.2 μM CcbF Y195G, combined under anaerobic conditions. Afterward, 18O2 gas (98%) or air was injected into the vial. For the H218O labelling experiment, 10 μl of H218O (97%) was added for the preparation of 25-μl reaction solutions (final concentration of H218O of 38.8%). The enzyme reactions were performed overnight at 20 °C and quenched by adding 100 μl of methanol. The samples were stored at −20 °C until LC–MS analysis to decrease the exchange rate with solvents. The products were analysed with the same method as that for the standard reaction products.
For the deuterium labelling experiments of LmbF and its variant, 15.7 μl of D2O (99.8%) was added for the preparation of 25-μl reaction mixtures (final concentration of D2O of 62.7%) containing 100 mM KPi, 0.5 mM EDTA, 0.5 mM DTT, 0.1 mM PLP, 40 μM substrate 4, and 0.1 nM wild type or 2 μM mutant LmbF. After incubation for 5 min at 37 °C, the reactions were quenched by adding 100 μl of methanol. For the deuterium labelling experiments of CcbF and its variants, 15.7 μl of D2O (99.8%) was added for the preparation of 25-μl reaction mixtures (final concentration of D2O of 62.7%) containing 100 mM KPi, 0.5 mM EDTA, 0.5 mM DTT, 0.3 mM SAM, 0.1 mM PLP, 40 μM substrate 4, 10 μM LmbG and 4 μM wild type or mutant CcbF. After incubation for 120 min at 37 °C, the reactions were quenched by adding 100 μl of methanol. Reactions in 100% H2O were performed as negative controls. The samples were analysed with the same method as that for the standard reaction.
Production and purification of the natural thiooctose substrate of LmbF and CcbF (4)
The seed culture of the S. lincolnensis ΔlmbIH strain17 was prepared by inoculating spores into 50 ml of Yeast Extract-Malt Extract (YEME) medium in 500-ml flat-bottom boiling flasks, which were incubated at 28 °C for 30 h, 180 r.p.m. A 2-ml portion of the seed culture was then inoculated into 40 ml of avermectin medium (AVM) medium in 500-ml flat-bottom boiling flasks, and incubated at 28 °C for 120 h. The supernatants from 30 flat-bottom boiling flasks were used in the next steps. The cells were centrifuged at 4,000g at 4 °C for 10 min, and the supernatant was stored at −20 °C. The compound of interest was extracted in two steps. First, a glass column containing Amberlite XAD-4 was used, and the amount of the sorbent was ~5 cm in diameter and 10 cm in height. Methanol (MeOH) followed by water was used to equilibrate the column before loading the supernatant, which was adjusted to pH 9–10 with ammonium hydroxide. The sorbent was then washed with 1 mM ammonium formate (pH 9). Absorbed compounds were subsequently eluted with a methanol–water solvent system: from methanol–water 10:90 (vol/vol) up to 80:20 (vol/vol) in steps of 10% and a 100-ml volume of each solvent. Fractions including the compound of interest were collected, pre-concentrated and adjusted to pH 2–3 with formic acid. Next, MCX 35 cc (6 g) (Waters) cartridges were used. The cartridge was conditioned and equilibrated with MeOH followed by 2% formic acid, then the eluate collected from the previous extraction was applied to the cartridge, which was washed with 2% formic acid and MeOH. Thereafter, 200 ml of MeOH with 5% of an aqueous solution of ammonium hydroxide (29%) was loaded to elute the compound of interest. For further purification of the natural substrate, a two-step preparative scheme was employed. The first step was performed with a Triart Prep C18-S column (20 × 250 mm, 5 μm; YMC) using a two-component mobile phase: A, 0.1% formic acid and B, methanol. Elution was performed at a flow rate of 3.5 ml min−1 with a linear gradient (min, % of B: 0 min, 5%; 27.5 min, 63%; 28 min, 100%; 43 min, 100%), followed by 15 min of equilibration with 5% solvent B. The second step was performed on an Xterra Prep RP18 column (7.8 × 150 mm, 5 μm; Waters) using a two-component mobile phase: A, 1 mM ammonium formate (pH 9.0) and B, CH3CN. The flow rate was 0.7 ml min−1 with a gradient of 10% to 50% B, for 60 min. Fractions containing the natural thiooctose substrate were monitored using an Acquity UPLC system with a 2996 PDA detection system (194–600 nm), connected to an LCT Premier XE time-of-flight mass spectrometer (Waters). The sample was loaded onto an Acquity Premier BEH amide LC column (2.1 × 50 mm, 1.7 µm), maintained at 40 °C. A two-component mobile phase—solvents A (20 mM ammonium formate (pH 4.75):CH3CN = 1:1 vol/vol%) and B (20 mM ammonium formate (pH 4.75):CH3CN = 1:9 vol/vol%)—was used for separation. Elution was performed at a flow rate of 0.4 ml min−1 with the following gradient (min, % of B: 1 min, 99%; 7 min, 1.0%; 9 min, 1.0%; 10 min, 99%), followed by 2 min of equilibration with 99% solvent B.
Large-scale reaction for structural determination of 9
The large-scale reaction of the CcbF Y195G variant with substrate 4 was performed in a 5-ml reaction mixture containing 100 mM KPi (pH 8.0), 400 μM substrate 4, 0.5 mM EDTA, 0.5 mM TCEP, 0.5 mM PLP and 20 μM enzyme, incubated overnight at 37 °C. After incubation, the reaction was quenched with an equal volume of methanol. For isolation of product 8, the reaction was purified by HPLC with an LC-20AD and SPD-M20A system (Shimadzu Corporation) equipped with an XSelect HSS T3 OBD Prep column (10 × 250 mm, 5 μm; Waters). Gradient elution was performed with solvent A (NH4OH, pH 9.0) and solvent B (CH3CN) at a flow rate of 3 ml min−1 (T = 0 min, 20% B; T = 5 min, 20% B; T = 20 min, 100% B; T = 25 min, 100% B; T = 25.5 min, 20% B). Finally, 0.5 mg of compound 9 was obtained from 1 mg of 4. 1H NMR (900 MHz, CD3OD): δ 5.45 (d, J = 5.5, 1H), 4.32 (d, J = 6.8, 1H), 4.14 (m, 2H), 4.05 (d, J = 3.0 Hz, 1H), 3.97 (dq, J = 6.4, 6.3 Hz 1H), 3.62 (dd, J = 3.0,10.1 Hz, 1H), 3.31 (d, J = 14.8 Hz, 1H), 3.21 (dd, J = 6.0, 8.8 Hz, 1H), 3.20 (d, J = 14.8 Hz, 1H), 2.95 (dd, J = 4.3, 10.6 Hz, 1H), 2.38 (s, 3H), 2.25 (m, 1H), 2.07 (dd, J = 8.8, 9.9 Hz, 1H), 2.00 (m, 1H), 1.81 (m, 1H), 1.32–1.38 (m, 4H), 1.22 (d, J = 6.3 Hz, 1H), 0.94 (t, J = 5.6 Hz, 1H), 13C NMR (225 MHz, CD3OD): 176.9, 173.6, 86.5, 70.4, 69.3, 69.3, 68.7, 67.9, 66.8, 62.4, 55.1, 40.4, 37.5, 37.4, 35.5, 32.2, 21.3, 18.6, 13.2. ESI-HRMS calculated for C19H35N3O7S [M + H]+ = 450.2268, found: 450.2286.
Crystallization and structure determination
Crystals of LmbF and CcbF were obtained after one day at 20 °C. All crystals were obtained using the sitting-drop vapour-diffusion method with the following reservoir solutions: LmbF, 0.1 M MES, pH 6.5, 27% PEG 4000, 0.8 M LiCl; CcbF, 0.1 M Tris-Cl, pH 8.5, 31% PEG 4000, 0.16 M NaOAc. The crystals were transferred into cryoprotectant solution (reservoir solution with 25% (vol/vol) glycerol), then flash-cooled at −173 °C in a nitrogen-gas stream. The X-ray diffraction datasets were collected at BL-1A (Photon Factory), using a beam wavelength of 1.1 Å. Diffraction datasets were processed and scaled using the XDS program package52 and Aimless in CCP4 (ref. 53). The initial phases of the LmbF and CcbF structures were determined by molecular replacement, using the model structures of LmbF and CcbF, constructed by ColabFold54, as the search models, respectively. Molecular replacement was performed with Phaser55 in PHENIX56. The initial phases were further calculated with AutoBuild in PHENIX56. The structures were modified manually with Coot57 and refined with PHENIX.refine58. The final crystal data and intensity statistics are summarized in Extended Data Table 1. The Ramachandran statistics are as follows: 98.4% favoured, 1.6% allowed for LmbF; 97.9% favoured, 2.1% allowed for CcbF. A structural similarity search was performed, using the Dali program server59. All crystallographic figures were prepared with PyMOL (DeLano Scientific, http://www.pymol.org).
Docking and MD simulations of LmbF and CcbF
Despite the availability of numerous docking simulation software tools, such as Autodock Vina60, the accurate prediction of protein–ligand complex coordinates is challenging, especially when only the apo form is available. This difficulty arises mainly because the software tools cannot consider the conformational changes of the main and side chains between the apo and holo forms. Meanwhile, in our study, the predicted coordinates of the PLP moiety of the ligand should not deviate substantially from those observed in our LmbF and CcbF crystal structures. To circumvent this, we performed only MD-based energy minimization and structural relaxation steps to generate the initial structure of the LmbF/CcbF–ligand complex after docking the ligand manually, instead of conventional docking simulations.
To obtain parameters for MD simulations, the structures of the external aldimine intermediate and the quinonoid intermediate of substrate 4 were optimized using Gaussian 16 Rev. C.02 (ref. 61) at the B3LYP level of theory62,63,64,65 with the 6-31+G(d,p) basis set. Their partial charges were obtained by restrained electrostatic potential (RESP), using the ANTECHAMBER66 module implemented in AmberTools22 (ref. 67). The general AMBER force field 2 (GAFF2)68 was used for the mechanical parameters of the ligands.
Ligand docking into the crystal structures was conducted manually according to the following procedure. First, the pyridine ring moiety of the external aldimine intermediate was superimposed on the position of the one observed in the respective crystal structures of LmbF and CcbF. The orientation of the Cα–H bond of the cysteine moiety in LmbF was arranged as shown in Fig. 3. Second, considering the voids present at the dimer interface, both dihedral angles −90° or −180° are possible for N–Cα–Cβ–S (Supplementary Fig. 11). However, we adopted −90° as −180° is unfavourable for the reaction, because the distance between the ε-amino group of Lys270 and the S atom was elongated. The rest of the ligand was modelled using the optimized substructure obtained by Gaussian 16. In CcbF, in addition to this conformation, the intermediate was docked so that the Cα–COO– bond was perpendicular to the pyridine ring, as shown in Fig. 4.
After adding H atoms, the modelled LmbF and CcbF complexes were fully solvated in the OPC water model69 in a cubic periodic box, then neutralized by adding Na+ and Cl– ions via the Amber LEaP module70. The ff19SB force field71 was used for the protein. Short-range van der Waals and electrostatic interactions were cut off beyond 10 Å, and the particle mesh Ewald (PME) method72 was used for long-range electrostatic interactions. The system was first relaxed using 200 steps of the steepest descent minimization, with a 1,000 kcal mol−1 Å−2 constraint applied to the heavy atoms of the protein. Subsequently, the entire system was subjected to 200 steps of the steepest descent minimization without restraints. Next, to gradually heat the system, 1-ns MD simulations were performed at 300 K and 1.0 × 105 Pa under the NPT ensemble. During equilibrations, the SHAKE algorithm73 was used to constrain the bonds including H atoms, and the integration time step was set to 2 fs. The Berendsen weak coupling algorithm74 was used to maintain constant temperature and pressure. We repeated the minimization and equilibration steps twice to resolve steric clashes between the ligand and the protein in the initial modelled structure, especially around LmbF-Leu83 and -Trp150. After equilibration, 50-ns conventional production runs were performed. Equilibrations and production runs were carried out using the PMEMD module75 of AMBER 22 (ref. 67). The distance and dihedral calculations were carried out using CPPTRAJ76.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.






留言 (0)