Ethics statement and animal use
All animal experiments were conducted in accordance with the approved animal care guidelines outlined in the European Communities Directive 2010/63/EU. These experiments were conducted with the authorisation of the State Veterinary and Food Administration in Bratislava (2978-5/2021 − 220 and 4518/19–221/3) under the supervision of the Ethical Council of Neurobiology BMC SAS. Wistar albino normal-weight rats (250–300 g, obtained from Velaz Ltd., Czech Republic) and ZDF rats (350–450 g, obtained from Dobrá Voda, Slovakia) were housed in standard conditions (20 ± 2 ºC, 55% ± 5% relative humidity and a 12-h light/dark cycle, and were provided food and water ad libitum. The experiments were conducted with 12-week-old male rats to minimise the influence of non-modifiable risk factors for stroke, such as older age and sex. All efforts were taken to minimise suffering.
Experimental design
Each experimental group contained 10 animals, although the minimum number of animals per group was set at 6 due to spontaneous death caused by cerebral ischemia as calculated previously (Bonova et al. 2016, 2020). The control group was set to 6 rats.
The study was divided into two sub-studies. For the first sub-study, the effect of a second round of RIPostC in ZDF rats was evaluated. Prior to surgery, rats were randomly assigned to one of the following four groups: control, ischaemia, tolerant and double tolerant. Rats from the ischaemia, tolerant and double-tolerant groups were subjected to 90 min of middle cerebral artery occlusion (MCAO) using an intraluminal filament technique developed by Longa et al. (Longa et al. 1989). The tolerant rats were subjected to one round of RIPostC, consisting of three cycles of 5 min of ischaemia– induced by placing an elastic rubber band tourniquet around the hind limb – followed by 5 min of reperfusion. This procedure was performed within 1 h after MCAO. A second round of the same RIPostC protocol was administered within 1 h after the previous IR cycle to the double-tolerant rats.
For the second sub-study, a tolerant phenotype in ZDF rats was induced by administering the secretome derived from blood cells of lean, healthy Wistar rats subjected to one round of RIPostC (Tolerant secretome group). The blood cell–derived secretome of the tolerant animals was prepared as described below and then intravenously injected into the ZDF rats within 30 min after transient ischaemia (90 min of MCAO). The effect of tolerant secretome was compared with non-tolerant secretome derived from blood cells of lean, healthy Wistar rats non-subjected to RIPostC (Non-tolerant group). The control rats represented intact ZDF rats (Control group). The rats were euthanised 24 h after the surgery under deep anaesthesia induced by 4% isoflurane.
Middle cErebral Artery Occlusion (MCAO) model
MCAO induction followed the procedure described by Longa et al. (Longa et al. 1989). The rats were anaesthetised using 4% isoflurane. A commercially available 6 − 0 monofilament nylon suture, which had a silicone rubber-coated tip, was inserted through an incision made in the external carotid artery into internal carotid artery to occlude the origin of the right middle cerebral artery (MCA) for 90 min. To effectively block the MCA, the filament was inserted approximately 19–20 mm from the carotid bifurcation. Cerebral blood flow was monitored using a laser-Doppler flow meter (Periflux System 5000, Perimed AB, Sweden). A 407 probe, along with an appropriate holder, was placed on the skull over the MCA (5 mm lateral and 1 mm posterior to bregma). Only rats whose blood flow dropped below 80% following filament insertion were included in the study.
Remote ischemic postconditioning
RIPostC was performed within 60 min after MCAO. The rats were subjected to three cycles of 5 min of ischaemia followed by 5 min of reperfusion of the femoral artery by placing an elastic rubber band tourniquet around the hind limb in a tight position to occlude the arterial blood supply.
Preparation of the blood cell–derived secretome
The blood of the RIC-stimulated (tolerant) and unstimulated (non-tolerant) Wistar rats was collected by cardiac puncture and processed as it was described previously (Bonova et al. 2016, 2020). After centrifugation (4500 g, 15 min, 4 °C), the blood plasma was discarded. To retain the original blood volume, the pellet, which represented a mix of all blood cell types, was washed and resuspended in artificial plasma (hydroxyethyl starch [HES] 130/0.4 in an isotonic electrolyte solution, 6% Volulyte, Fresenius Kabi Deutschland GmbH). Subsequently, the samples were incubated in a CO2 incubator (37 °C for 150 min) followed by centrifugation (4500 g, 15 min, 4 °C). The supernatant was removed and centrifuged again (15,000 g, 15 min, 4 °C) to remove any remaining cells and cellular debris. The final supernatant – the blood cell–derived secretome with preserved exosomes and microparticles – was injected intravenously into ZDF rats that had undergone 90 min of MCAO according to Longa et al. (Longa et al. 1989), as mentioned previously.
Brain sample preparation
After decapitating the animals, as described previously, their brains were rapidly removed and cut into 2-mm coronal slices, which were used for 2,3,5-triphenyltetrazolium chloride (TTC; Sigma), FluoroJade B (FJB; Histo-Chem Inc., USA) and haematoxylin & eosin (H&E) staining. For the tissue analysis of glutamate, slices were dissected from the ischaemic core and penumbra of the ipsilateral and contralateral hemispheres, as described previously (Ashwal et al. 1998), with minor modifications. Subsequently, the tissue was weighed, homogenised in homogenisation buffer (20 mM Tris-HCl pH 7.5 containing 1 mM DTT, 50 mM magnesium acetate, 140 mM KCl, 1 mM EDTA, 2 mM EGTA with the addition of Protease Inhibitor Cocktail Tablets, Roche, Germany) and centrifuged at 12,000 rpm for 15 min at 4 ºC. The total protein concentration was determined using the method described by Bradford (Bradford 1976), using a standard curve established with bovine serum albumin (BSA). The core and penumbral post-mitochondrial supernatants were precipitated with 1 M ice-cold perchloric acid (PCA; 1:20; 10 min, 4 ºC), and centrifuged (12,000 rpm, 10 min, 4 ºC). The supernatant was collected and stored at −80 ºC for later analysis.
Blood sample processing
Whole blood samples were obtained by cardiac puncture and collection in heparin-coated tubes 24 h after the ischaemia. Blood samples were centrifuged at 12,000 rpm for 10 min at 4 ºC to separate plasma and blood cells. Catalase (CAT) activity was measured in the plasma. Superoxide dismutase (SOD) activity was measured in the blood cell lysate. The blood cell pellet was suspended in distilled water equivalent to the volume of plasma collected. After rapid freezing (10 min, −20 ºC) and defrosting with three cycles at 37 ºC, the samples were centrifuged at 12,000 rpm for 10 min at 4 ºC. The supernatant, representing the blood cell lysate, and plasma samples were stored at −80 ºC until further analysis.
Lymphocytes assigned for the comet assay were isolated from 100 µl of whole blood and diluted in phosphate-buffered saline (PBS) at a 1:4 ratio. Isolation was performed via density centrifugation (2000 rpm, 5 min, 4 ºC) using a Ficcol–Paque™ plus gradient. The layer containing lymphocytes was collected, rewashed in PBS (1:4), and directly used for assessing DNA via Single-Cell Gel Electrophoresis.
Glutamate was measured in the whole blood. Samples were deproteinised by adding ice-cold 1 M PCA at a 1:1 ratio, precipitated for 10 min on the ice and centrifuged at 12,000 rpm for 10 min at 4 ºC. The supernatant was collected and stored at −80 ºC for subsequent analysis.
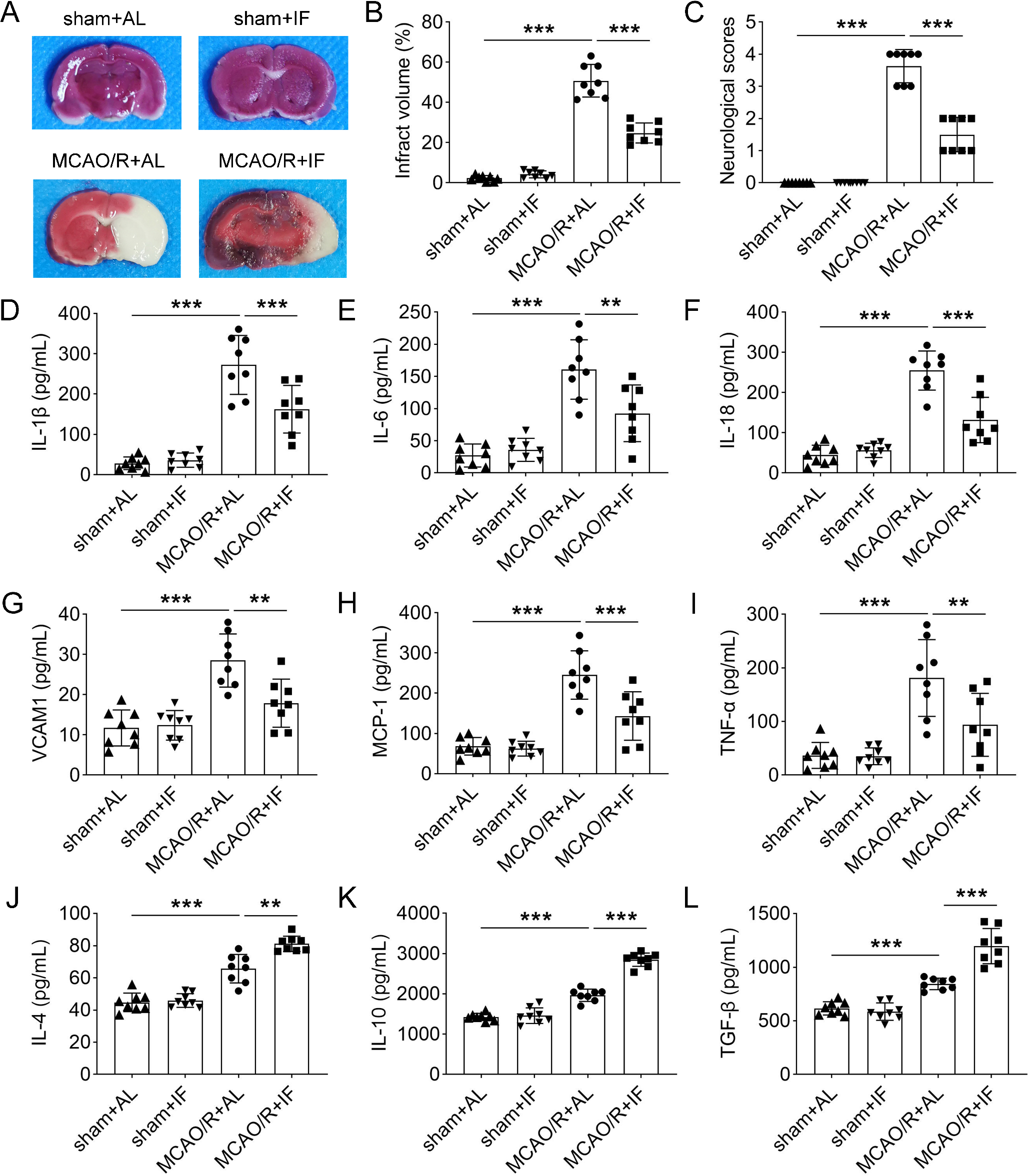
Evaluation of the infarct volume and neurological deficits
After dividing the brain into 2-mm-thick coronal sections, these sections were promptly placed in a solution consisting of 2% TTC in PBS at 37 ºC for 30 min for vital staining. Subsequently, they were immersed in a 10% formaldehyde solution for fixation for 24 h. Each section was scanned using a high-resolution scanner (Epson Perfection 4490 Photo, resolution 600 dpi), and digital images of five slices from each brain were analysed using Image J software by blinded investigator (version 1.8.0, National Institute of Health, Bethesda, MD, USA). The extent of brain damage was quantified by determining the ratio of the infract area (white area on the right side) to the area of the undamaged, contralateral hemisphere. The size of infarct was calculated using the following equation: infarct rate (mm3) = (non-ischaemic volume/ischaemic volume) × infarct volume (Callaway et al. 2000).
The animals were neurologically assessed 1 and 24 h after reperfusion. Evaluation and scoring was blind, without experimenter knowledge of rat’s membership in group. A four-point scale described by Bederson et al. (Bederson et al. 1986) was used for neurological assessment: (1) forelimb flexion and torso turning to the contralateral side when lifted by the tail; (2) the same behaviour as grade 1 and decreased resistance to a lateral push; (3) the same behaviour as grade 2 with unilateral circling; and (4) no spontaneous walking and a depressed level of consciousness. Each category was scored from 0 to 3 (the greater the score, the more severe the impairment). Rats with neurological deficits lower than 2 were excluded from the study.
Histology
After TTC labelling, the fixed brains were dehydrated in a 30% sucrose in PBS. They were sliced into 25-µm-thick coronal sections using a Leica CM1850 cryostat and mounted directly on gelatine-coated slides. The sections were allowed to air dry and were subsequently rehydrated in decreasing concentrations of ethanol (from 100 to 50%). Following rehydration, the sections were stained using the H&E Staining Kit (Abcam, Cambridge, UK). After staining, the tissue sections were rinsed with distilled water, dehydrated through a series of ethanol solutions (from 70 to 100%), cleared in xylene and mounted with mounting solution (DPX Mountant; Fluka Chemie AG, Switzerland). Once the mounting solution had solidified, images were captured using the Aperio AT2 digital scanner (Leica Biosystems, Wetzlar, Germany). Ischaemic damage was assessed and quantified as the infarct volume by measuring the areas of cell death, as described previously (Osborne et al. 1987), and by counting pyknotic cells in the striatum and cortex within the penumbra per mm2 using the ImageJ software.
Fluoro-Jade B staining
Twenty-five-micrometre-thick coronal sections were prepared following the same procedure as for H&E staining. The sections mounted on gelatine-coated slides, air-dried and then heated at 50 ºC for at least 15 min before staining. The slides were immersed in absolute alcohol, 70% ethanol and for 3 min in distilled water. The sections were then placed in a 0.06% potassium permanganate solution for 15 min and subsequently rinsed in distilled water for 2 min. Afterwards, the sections were transferred into a 0.0001% solution of FJB dissolved in 0.1% acetic acid for 30 min. The slides were washed three times with distilled water, left to air dry overnight at room temperature, cleared in xylene and then cover-slipped with DPX Mountant (Fluka Chemia AG). The slides were examined using an Olympus BX51 microscope equipped with an Olympus DP50 camera. FJB-positive neurons within the ischaemic penumbra were counted in 10 randomly selected 1-mm2 areas and expressed per mm2 of tissue using the ImageJ software.
Glutamate concentration
The glutamate concentration in blood and brain tissue was measured by using a modified enzymatic fluorimetric method described by Graham and Aprison (Graham and Aprison 1966; Kravcukova et al. 2009). The method is based on the fluorimetric detection of NADH produced by the glutamate and NAD + reaction catalysed by glutamate dehydrogenase. The glutamate concentration in the reaction is directly proportional to the NADH concentration. Briefly, 10 µl of supernatant, 190 µl of reaction buffer (0.25 M hydrazine hydrate/0.3 M glycine buffer, pH 8.6) containing 200 nM NAD + and 15 U of glutamate dehydrogenase were pipetted into a black 96-well plate. The fluorescence intensity of the final product (NADH) was measured using a Synergy™ 2 Multi-Mode Microplate Reader (BioTek) at 460 nm and an excitation wavelength of 360 nm 30 min after incubation at room temperature. The glutamate blood concentration is expressed as µmol/l blood, and the glutamate brain concentration was normalised according to the protein content and is expressed as µmol/mg protein.
Comet assay
The alkaline comet assay was performed based on the method described by Singh et al. (Singh et al. 1988), with minor modifications. First, microscope slides were covered with 1% normal-melting-point agarose in PBS (pH 7.4) and allowed to dry at room temperature. A suspension of lymphocytes was mixed with 1% low-melting-point agarose in PBS (pH 7.4). This mixture was spread onto slides precoated with agarose, covered with coverslips and allowed to dry in a refrigerator for 2 min. The coverslips were then removed, and the slides were immersed in a lysis solution (containing 2.5 mol/l NaCl, 100 mmol/l Na2EDTA, 10 mmol/l Tris, 1% Triton X-100 and 10% DMSO, pH 10) for 1 h at 4 ºC. Subsequently, the slides were transferred to an alkaline electrophoresis buffer (comprising 5 mol/l NaOH and 200 mmol/l Na2EDTA, pH 13) for 20 min at 4 ºC, and electrophoresis was performed at 25 V for 25 min at 4 ºC. Then, the slides were neutralised in a neutralisation buffer solution (containing 400 mmol/l Tris, pH 7.5) for 15 min at 4 ºC. After air-drying, DNA was stained with SYBR Green, and 100 cells on each slide were examined under a fluorescence microscope (Olympus BX51, excitation filter at 485 nm, emission filter at 520 nm) equipped with a camera (Olympus DP50). The images were analysed using the Comet Score™ v1.5 image analysis system (TriTek Corp., USA). DNA damage was assessed based on the parameter ‘% DNA in tail’, calculated as the difference between 100% of cell fluorescence intensity and the intensity within the head region, and represented as a percentage.
Determination of antioxidant enzyme activitiesSuperoxide dismutase
Superoxide dismutase (SOD) activity was measured according to the method described by Sun et al. (Sun et al. 1988). The standard assay substrate mixture contained the following (in 200 µl): 1 M xanthine (Sigma), 0.1 M EDTA, 5.6 × 10−2 M p-nitrotetrazolium (NBT, blue grade III, Sigma-Aldrich, Steiheim, Germany) and 1 M BSA (Fluka) in 0.1 M sodium phosphate (pH 7.8). This assay employs xanthine-xanthine oxidase as a source of superoxide to prevent the reduction of NBT by superoxide. It quantitatively measures the absorbance of NBT, which is converted into a blue formazan compound by superoxide, at 560 nm and room temperature. SOD in the sample breaks down superoxide, slowing the production of blue formazan. In the context of blood cells, one unit of SOD activity is defined as the amount that reduces the absorbance change by 50%.
Catalase
Catalase (CAT) activity in erythrocytes was quantified using spectrophotometry, following the method described by Góth (Góth 1991). This method is based on the creation of a stable complex between hydrogen peroxide and ammonium molybdate. In brief, 20 µl of the sample was mixed with 100 µl of the substrate (65 µmol/ml of hydrogen peroxide in a 60 nmol/l sodium potassium phosphate buffer at pH 7.4) for 60 s at 37 ºC. A stop solution, comprising ammonium molybdate (32.4 mmol/l; 100 µl), was used, and the yellow complex formed by molybdate and hydrogen peroxide was measured at 405 nm. One unit of CAT activity is defined as the quantity of enzyme required to break down 1 µmol of hydrogen peroxide within 1 min under these specific conditions.
Glutathione peroxidase
Glutathion peroxidase (GSH) activity was measured in the whole blood sample using the colorimetric method described by Alisik et al. (Alisik et al. 2019), with minor modifications. A sample of precipitated blood was incubated with 10 mM Ellman’s reagent in 500 mM Tris buffer (pH 8.2) with 10 mM EDTA for 10 min at room temperature. The absorbance was measured at 415 nm after the incubation. Glutathione (0–15 nM) was used as a calibrator for the assay.
Statistical analysis
Data were analysed and plotted using GraphPad Prism (Graph-Pad Software, San Diego, CA, USA). Statistical analysis was performed using one- and two-way analysis of variance, followed by the Dunnett post hoc test. The criterion for statistical significance was p < 0.05.





留言 (0)