
記住我

All experimental protocols involving mice were approved by the University of Queensland Animal Ethics Committee. WT (C57BL/6J, or littermate controls as appropriate), Casp1C284A/C284A (B6J-Casp1C284Aem1Ksc CR; generated in-house32), Casp11−/− (B6.Casp4tm; backcrossed to C57BL/6J)47, Gsdmd−/− (C57BL/6N-Gasdmdem1Vmd; backcrossed to C57BL/6J)6, Ninj1−/− (C57BL/6N-Ninj1-ENU-KO)8 and Clec9a-GFP knock-in (Jackson, B6(Cg)-Clec9atm1.1Crs/J)1 mice were housed in specific pathogen-free conditions at the University of Queensland. Six to 20 week-old male and female animals were sex- and aged-matched for each experiment.
Molecular cloning, plasmid constructs and mRNAsThe pMSCV-PLCδ-PH-GFP construct10 was expressed in primary bone marrow cells by retroviral transduction23. The coding sequences of pEF6-mCLEC9A(WT)-V5 and pEF6-mCLEC9A(Y7F)-V5 were synthesized by Integrated DNA Technologies and inserted into a modified pEF6 backbone. All plasmid open reading frame sequences are supplied in the Supplementary Data.
Cell death effector-encoding messenger RNAs (mRNAs), corresponding to the constitutively active portion of murine GSDMD (residues 1–276), GSDME (residues 1–270), GSDMA3 (residues 1–262), tBID (residues 60–195) and MLKL (residues 1–125, 4HB domain) were synthesized and in vitro transcribed with 5-methoxyuridine modification by the University of Queensland BASE Facility. All mRNAs were validated as endotoxin free. All mRNA open reading frame sequences are supplied in the Supplementary Data.
Cell cultureAll cell culture work was conducted under sterile conditions in class II biosafety cabinets and cells were incubated at 37 °C with 5% CO2. Cell lines were routinely tested for mycoplasma contamination and maintained mycoplasma-free. The murine macrophage cell line RAW264.7 (American Type Culture Collection TIB-71) was maintained in full RAW264.7 media (Roswell Park Memorial Institute, RPMI, 1640 medium (cat. no. 21870092, Gibco), 5% endotoxin-free fetal calf serum (FCS; Gibco), 10 U ml−1 penicillin–streptomycin (cat. no. 15070063, Gibco), 2 mM GlutaMAX (cat. no. 35050061, Gibco)). The retroviral packaging Platinum-E (Plat-E) cell line48 was maintained in full Plat-E media (Dulbecco’s Modified Eagle Medium (DMEM; cat. no. 11995073, Gibco), 10% FCS, 10 U ml−1 penicillin–streptomycin, 10 µg ml−1 blasticidin and 1 µg ml−1 puromycin).
Primary murine bone marrow-derived macrophages (BMDMs) were differentiated from bone marrow progenitors49 and used for experiments on day 7 or 8 of culture. Primary macrophages were maintained in full BMDM media (RPMI, 10% FCS, 10 U ml−1 penicillin–streptomycin, 2 mM GlutaMAX, 150 ng ml−1 recombinant human colony-stimulating factor-1 (CSF-1; endotoxin free, expressed and purified by the University of Queensland Protein Expression Facility)). cDC1s were produced from fresh bone marrow progenitors differentiated in full cDC1 media (Iscove’s Modified Dulbecco’s Medium (IMDM) medium (cat. no. 12440053, Gibco), 10% FCS, 10 U ml−1 penicillin–streptomycin, 2 mM GlutaMAX, 10 mM HEPES (cat. no. 15630080, Gibco), 1 mM sodium pyruvate (cat. no. 11360-070, Gibco), adjusted to 308 mOsm and supplemented with 200 ng ml−1 recombinant mouse Flt3-ligand (Flt3L; 250-31L, Peprotech, or 130-097-372, Miltenyi Biotech)) and used for experiments on day 8 of culture.
Human monocyte-derived macrophages (HMDM) were produced from buffy coats from blood donations to the Australian Red Cross Blood Service from anonymous, informed and consenting adults. Human peripheral blood mononuclear cells were isolated from screened buffy coats by density centrifugation with Ficoll-Plaque Plus (GE Healthcare), and monocytes were isolated by magnetic-assisted cell sorting49. Monocyte-derived macrophages were generated by differentiation over 7 days with 150 ng ml−1 recombinant human CSF-1 at 37 °C with 5% CO2. HMDMs were used for experimentation on day 7 of their differentiation.
Immortalized HBECs were maintained in complete keratinocyte media (Gibco, cat. no. 17005042, supplemented with the provided bovine pituitary enzyme and epidermal growth factor) and passaged at 70–80% confluency. Then 5 × 104 cells were seeded onto glass coverslips in complete keratinocyte media 24 h before CASP4 activation assays.
Inflammasome and apoptosis stimulationMurine or human macrophages were plated at 0.1 × 106 cells per 0.5 ml in full media in 24-well cell culture-treated plates containing a 2.5 cm (1 inch) coverslip (for microscopy) or 0.1 × 106 cells per 0.1 ml full media in 96-well cell culture-treated plates (for lactate dehyrogenase (LDH) assay). To activate the CASP11 inflammasome, cells were primed with 1 µg ml−1 of PAM3CSK4 (tlrl-pms, InvivoGen) for 16 h. Media was then removed and replaced with Opti-MEM (cat. no. 31985062, Life Technologies) supplemented with 150 ng ml−1 CSF-1. Next, 2 µg ml−1 of ultrapure Escherichia coli K12 LPS (tlrl-peklps, InvivoGen) was prepared for cytosolic delivery with FuGENE HD (cat. no. E2311, Promega) and the FuGENE HD/LPS complex was delivered to the cells by centrifugation at 600g for 10 min at room temperature. Cells were then incubated for a further 2 h or as otherwise stated. To activate NLRP3, full media was removed and macrophages were primed with 100 ng ml−1 of ultrapure E. coli K12 LPS in Opti-MEM supplemented with 150 ng ml−1 CSF-1 for 4 h, before exposure to 5 µM (BMDM) or 10 µM (HMDM, bone marrow-derived dendritic cells (BMDCs)) nigericin (N7143, Sigma Aldrich) for a further 30–45 min (or as indicated). The NAIP-NLRC4 inflammasome was activated using the protective antigen-Tox system that delivers proteins to the cytosol by N-terminal fusion to the Bacillus anthracis lethal factor (LFn), and transmembrane transport through the anthrax protective antigen channel50,51. For murine cells, we used a recombinant fusion protein of LFn with Legionella pneumophila flagellin (Fla1). For activation of human NAIP-NLRC4, we used a fusion of LFn and Salmonella typhimurium PrgI. Cells were stimulated with 250 ng ml−1 recombinant PA plus 125 ng ml−1 Fla1-LFn or 20 ng ml−1 PrgI-LFn in Opti-MEM supplemented with 150 ng ml−1 CSF-1 for 1 h. For sublytic inflammasome signaling, BMDM or BMDC were primed with LPS as before, and stimulated with 100 μg ml−1 PGPC (10044, Cayman; freshly reconstituted13). For induction of apoptosis, BMDMs were left unprimed and intrinsic apoptosis was induced with ABT-737 and S63845 (cat. nos. 11501 and 21131, Cayman Chemical, both used at 500 nM) for 2 h.
To activate the CASP4 inflammasome in HBECs, cells were primed with 1 µg ml−1 of PAM3CSK4 for 16 h and transfected with LPS as above, using the transfection reagent Lipofectamine LTX with PLUS reagent (cat. no. 15338100, Invitrogen). Cells were fixed 5.5 h after transfection for confocal imaging.
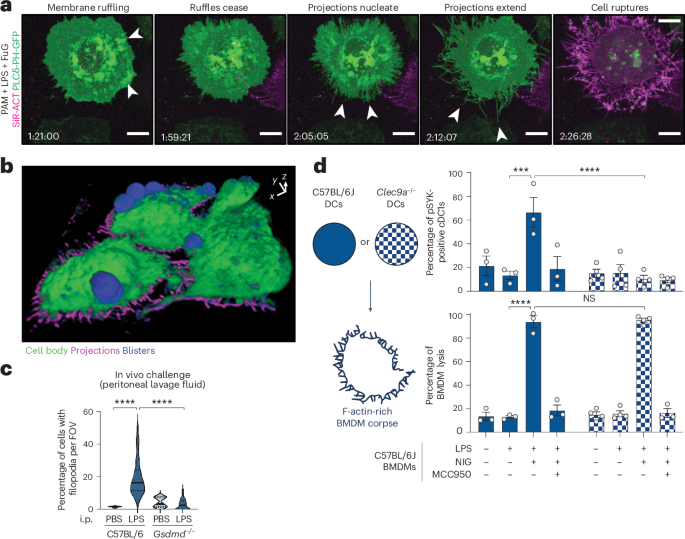
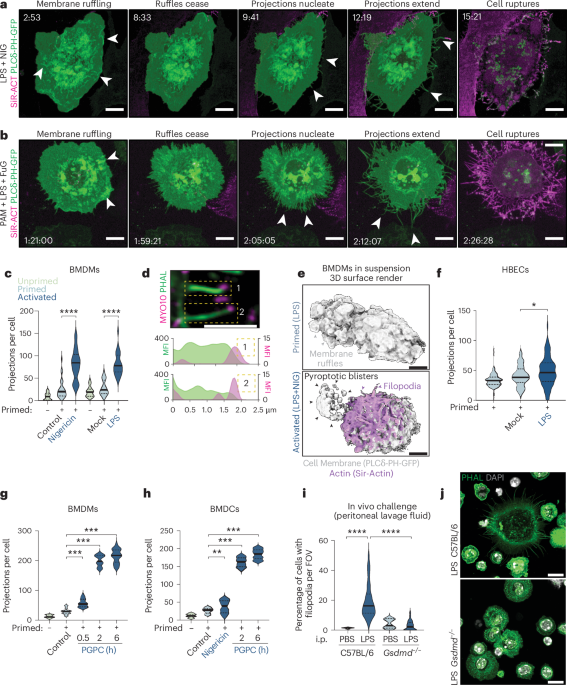
SEMSEM was performed on cells plated on 35-mm dishes with inbuilt high grid-500 1.5′ coverslips (cat. no. 81168, Ibidi). Following inflammasome stimulation, cells were fixed in 4% paraformaldehyde (PFA) (C004, ProSciTech) and 0.25% EM-grade glutaraldehyde (C002, ProSciTech) for 20 min. Cells were stained with Alexa Fluor 488 Phalloidin (6.6 µM, cat. no. A12379, Invitrogen) for 15 min and cells of interest were mapped in PBS using a Zeiss AxioObserver widefield microscope and ×40 (numerical aperture (NA) 1.3) or ×63 (NA 1.4) oil objectives using Zen Blue software. Cells were washed in PBS and refixed in 2.5% glutaraldehyde (C002, ProSciTech) in PBS in a BioWave microwave (Ted Pella Inc.) at 80 W for 3 min under vacuum. Dishes were subsequently washed in PBS and water then postfixed in 1% aqueous Osmium tetroxide (OsO4) at 80 W cycling for 6 min under vacuum. Fresh OsO4 solution was added, and cells were postfixed again in the BioWave. Dishes were washed twice in ddH2O and then serially dehydrated in increasing percentages of ethanol (30, 50, 70, 90, 100, 100%). Coverslips were separated from the dishes while submerged in 100% ethanol and transferred to a Tousimis Autosamdri-815 for critical point drying. Dry coverslips were mounted onto aluminum SEM stubs and carbon coated under vacuum in a Safematic CCU-010 Carbon coater. Stubs were loaded into a field emission FEI Scios Focus Ion Beam Scanning Electron Microscope, and regions of interest were correlated and imaged with secondary electron detection.
Fluorescence microscopyConfocal imaging was performed on cells fixed in 4% methanol-free formaldehyde (cat. no. 28906; Pierce, or 50-980-487; EMS) for 15–20 min at room temperature and stored at 4 °C in PBS overnight to quench remaining PFA until required, or quenched with 50 mM NH4Cl if processed immediately. All steps were carried out at room temperature, in the dark. Cells were labeled with 4,6-diamidino-2-phenylindole (DAPI) (0.1 µg ml−1, D9542, Sigma Aldrich) and Phalloidin-iFluor 405 (1,000×, cat. no. AB176752, Abcam), Alexa Fluor (AF) 488 Phalloidin (6.6 µM, cat. no. A12379, Invitrogen) or AF647 Phalloidin (13.2 µM, cat. no. A22287, Invitrogen). Antibodies used for immunolabelling were anti-ASC (1:200, N15; Santa-Cruz; 1:800, cat. no. 67824S, Cell Signaling Technology), anti-MYO10 (1:100, cat. no. AB224120, Abcam), antiphospo-SYK (1:100, cat. no. 2711S, Cell Signaling Technology), anticleaved CASP3 (1:100, cat. no. 9664S, Cell Signaling Technology) and anti-V5 (1:200, cat. no. AB27671, Abcam). Secondary antibodies used were goat anti-rabbit AF594 (cat. no. A32740, Invitrogen), donkey anti-mouse AF488 (cat. no. A21202, Invitrogen) and goat anti-rabbit AF647 (cat. no. A32733, Invitrogen). Cells were mounted onto slides using ProLong Gold antifade mounting media (cat. no. P36934, Invitrogen) and sealed with clear nail polish. Images were acquired on a Zeiss Axiovert 200 microscope stand fitted with a LSM 880 confocal scanner running Zeiss Zen Black software. The microscope was equipped with 405, argon ion, 561 and 633 nm lasers. Plan Apochromat ×40 (NA 1.3) or ×63 (NA 1.4) oil immersion objectives were used and where indicated, the 4Y-Fast Airyscan mode was used.
For analyses of filopodia formation in PGPC-treated cells, all steps were carried out at room temperature, in the dark and in PermQuench buffer (0.2% w/v bovine serum albumin and 0.02% w/v saponin in PBS). First, the specimens were permeabilized for 30 min and then stained with rabbit anti-ASC (1:100, cat. no. AG-25B-0006-C100, Adipogen) or anti-MYO10 (1:100, cat. no. AB224120, Abcam) for 1 h. Secondary anti-rabbit-AF488 (cat. no. R37116, Thermo Fisher Scientific) was used, and 1 μg ml−1 DAPI (cat. no. D1306, Thermo Fisher Scientific) and 1:400 AF647 Phalloidin (cat. no. A22287, Thermo Fisher Scientific). Specimens were washed and mounted on slides using ProLong Glass Antifade Mountant (cat. no. P36982, Thermo Fisher Scientific) and left to cure overnight. Imaging was performed using a LSM 880 scanning confocal microscope with fast Airyscan (4Y) controlled with Zeiss Zen Black software (Zeiss Instruments) and equipped with incident light fluorescence and laser illumination (405, 458, 488, 514, 561 and 633 nm), two GaAsP, two standard PMT NDD detectors and an Airyscan detector (32 GaAsP array). Imaging used a Plan Apochromat ×40/1.3 or a Plan Apochromat ×63/1.4 oil immersion objective (Zeiss Instruments). In a typical experiment, 9–25 fields of view were imaged using multi-position acquisition. Representative images were acquired using the Airyscan detector. Acquired microscopy data were processed using ImageJ/Fiji and filopodia were manually counted in a minimum of 40 cells per condition from randomly chosen fields of view.
Live fast Airyscan confocal microscopy imaged PLCδ-PH-GFP-expressing macrophages stained with SiR-actin23 incubated at 37 °C with 5% CO2.
For analysis of calcium oscillations, WT and Gsdmd−/− BMDMs were seeded in four-chamber 35-mm glass-bottom dishes (cat. no. D35C4-20-1.5-N, Cellvis) in CSF-1-supplemented phenol red-free Opti-MEM (cat. no. 11058021, Gibco), and primed for 3 h with LPS. To activate NLRP3, 5 μΜ nigericin was added together with the dyes SiR-Actin and the calcium biosensor Fluo-4 (cat. no. F10489, Invitrogen). Imaging commenced immediately.
For both fixed and live confocal microscopy, raw images were deconvolved over ten rounds of iterative deconvolution using Microvolution52 running in Fiji (Microvolution LLC). Images were further subjected to background subtraction and additionally, for quantification, a 2-pixel median filter. Images are represented as maximum intensity projections (MIPs) of Z-stacks, unless indicated otherwise in the figure legend. The number of projections per cell was quantified by manual counting by a blinded operator. Quantification of cell size in EGTA-treated BMDM was performed in Fiji by using the AutoThreshold plugin with the default method to create a mask of each cell. The surface area of each masked cell, or masked cell corpse fragment larger than 10 pixels (approximately 2 μm2), was then measured with the Analyse Particles function.
Lattice light-sheet imaged SiR-actin-stained primary macrophages expressing PLCδ-PH-GFP23. Coverslips (1.5′, 5-mm, cat. no. G400-05, ProSciTech) were placed in the 3i LLSM (v.2) 37 °C sample bath containing phenol red-free Opti-MEM and 5 µM nigericin. A selected field of view containing several PLCδ-PH-GFP-positive cells was imaged. A multi-Bessel beam optical lattice pattern was generated through a spatial light modulator and higher orders filtered through an annular mask to generate a noncontinuous light sheet that was dithered across the x axis. The sample was imaged by moving the sample stage in 0.495 µM increments across a region of ~75 × 50 × 100 μm using a 30-ms exposure time for each channel. The spatial light modulator was set to a sheet length of ~20 µM, 44 and 36 Bessel beams were used for the 488 and 642-nm channels, respectively. The inner and outer numerical aperture was 0.493 and 0.550 of the annular masks. The lattice light sheets were generated using the following settings: the spacing factor between each beam was 0.932 μm (488 nm) and 1.233 μm (642 nm), with a cropping factor of 0.175 (488 nm) and 0.190 (642 nm). Then, 161 Z slices were acquired at an angle of 32.8° relative to the sample and recorded using dual Hamamatsu Orca Flash 4.0 cameras. The composite three-dimensional (3D) images each took 12 s to acquire, inclusive of the set delay between sweeps and time taken to alternate between the 488 and 642-nm laser at each slice. Images were deconvolved and deskewed using Microvolution software before being exported to Arivis Vision4D (v.4.1.1) and Fiji/ImageJ (v.1.52k-1.54 f) for image processing. All acquisitions in Fig. 4a,b were subject to nonlinear adjustment (gamma v.2.0).
Imaging macrophages in suspensionBMDMs expressing PLCδ-PH-GFP were imaged in suspension by plating cells onto passivated glass-bottom dishes to prevent the cell from forming stable adhesions to glass. To PEG-passivated glass surfaces, four-chamber glass-bottom dishes (D35C4-20-1.5-N, Cellvis) were treated with poly-l-lysine (PLL, 20 kDa)-grafted with PEG (2 kDa) (PLL(20)-g[3.5]-PEG(2)) (SuSoS Surface Technology) resuspended in PBS at a final concentration of 1 mg ml−1. Glass-bottom dishes were passivated by overnight incubation at 37 °C and 5% CO2. The following day, the PLL-g-PEG solution was aspirated and 450 μl of suspension BMDM PLCδ-PH-GFP cells were seeded into each well with media supplemented with LPS and CSF-1 to prime for 4 h. Before imaging, the cell suspension in each well was pipetted up and down several times to encourage single cell suspension. To trigger pyroptosis, 50 μl of media containing LPS, CSF-1, SiR-actin and nigericin (5 µM) or vehicle was added to each well and imaging was immediately commenced. Imaging was performed on an inverted Andor Dragonfly 500 Spinning Disc Confocal microscope using a ×40 LWD Apochromat 1.15 NA water dipping objective and GenTeal Gel (Alcon) as an immersion medium, with air surrounding the objective sealed off to prevent the immersion medium from evaporating. To capture cells in suspension, temporal Z-stacks were acquired with an Andor Zyla 4.2 sCMOS camera over 45 min at intervals of 1–2 min within the Andor Fusion software. 3D cell surface renders were generated using Chimera X53.
Cell death assaysCell death was assayed by measuring LDH release, using the Cytotox96 nonradioactive cytotoxicity assay (Promega) or CyQUANT LDH Cytotoxicity Assays (cat. no. C20301, Thermo Fisher Scientific) and graphed as a percentage of total cell lysis (control cells treated with 0.1% Triton X-100 (cat. no. T9284, Sigma Aldrich) in Opti-MEM for 5 min to induce 100% cell lysis). Absorbance at 490 nm was measured using the Powerwave XS spectrophotometer (BioTek) or the Infinite PRO 200 plate reader (TECAN).
ImmunoblottingCell extracts were collected in boiling western lysis buffer (66 mm Tris-HCl pH 7.4, 2% SDS), and analyzed by SDS–PAGE and immunoblot25. Primary antibodies used were: α-v5 monoclonal clone Sv5-PK1 at 1:1,000 (cat. no. ab27671, AbD Serotec), α-GSDMD at 1:1,000 (cat. no. ab209845, Abcam) and glyceraldehyde 3-phosphate dehydrogenase polyclonal at 1:5,000 (cat. no. 2275-PC, R&D Systems). Secondary HRP-conjugated antibodies used were anti-rabbit IgG and anti-mouse IgG both at 1:5,000 (cat. no. 7074, 7076, Cell Signaling Technology). Secondary antibodies on membranes were inactivated by incubation with 30% hydrogen peroxide before reprobe. Membranes were visualized using a Fusion imaging system (Vilber).
Cell death induction by mRNA transfectionmRNA transfections were performed on days 6–7 of BMDM differentiation using the Neon transfection kit (cat. no. MPK1025R, Thermo Fisher). Briefly, cells were rinsed and lifted in PBS and concentrated to a cell density of 1.5 × 106 cells per 10 µl in Buffer R immediately before electroporation of 1 µg of GSDMD mRNA and molar ratio concentrations of the following death effectors: GSDME, GSDMA3, tBID and MLKL. Electroporator settings were 1,400 V, 20 ms−1, two pulses, using a 10-µl tip. After delivering the electric pulse, cells were transferred to 1.5-ml tubes containing prewarmed assay media (Opti-MEM + CSF-1) and plated at the desired cell density for imaging and LDH release quantification. Cells were allowed to recover for 1 h before LPS priming, and samples were collected over a time course of up to 24 h for analyses.
CLEC9A activationCLEC9A recognition of F-actin-rich pyroptotic corpses was first assayed by adding CLEC9A-expressing RAW264.7 cells to pyroptotic BMDMs. RAW264.7 cells stably expressing murine CLEC9A-V5 (WT or Y7F signal-deficient mutant) were cultured to 50% confluency the day before experimentation. Primary macrophages were stimulated to induce pyroptosis in 24-well plates with coverslips and washed three times in sterile Dulbecco’s PBS (cat. no. 14190144, Gibco) to remove inflammasome agonists, media and soluble cell and corpse components. A 30 mM pervanadate stock solution was prepared by diluting sodium orthovanadate in sterile Dulbecco’s PBS, followed by the addition of 30% H2O2 (cat. no. HA154-500M, Chem Supply Australia) to a final concentration of 0.18% (w/w) and incubated for 15 min at room temperature. 0.2 × 106 RAW264.7 cells in Opti-MEM containing 50 μM pervanadate were added to each well containing macrophages, centrifuged (500g, 5 min), and incubated for 30 min. p-SYK/mCLEC9A-V5 interactions were assayed using PLA (DUO92004, DUO92002 and DUO92008, Duolink In Situ PLA, Sigma Aldrich) according to the kit instructions, omitting the antibody preincubation step to reduce nonspecific background. Samples were imaged using confocal microscopy. RAW264.7 cells were manually segmented and subjected to background subtraction, median filter and MIP. Data are represented as the number of PLA puncta per cell.
CLEC9A recognition of F-actin-rich pyroptotic corpses was also assayed by culturing murine dendritic cells with pyroptotic corpses. WT, Gsdmd−/−, Ninj1−/− macrophages were plated at 0.25 × 106 cells per 0.5 ml in full BMDM media in 24-well cell culture-treated plates, and primed and stimulated as described above, with and without 1 h of pretreatment with 10 µM EGTA-AM before nigericin treatment for 45 min. Macrophage media was aspirated and assayed for LDH release, while adherent cells and corpse remnants were washed twice with prewarmed PBS in the plate. Next, 0.75 × 106 Flt3L-derived BMDCs (WT versus Clec9a−/−) were added to each well in 250 µl Opti-MEM containing 1 mM pervanadate, and incubated for 30 min before flow cytometric analyses.
Dendritic cell activationFor analyzing cDC1 dendritic cell activation by pyroptotic corpses, immortalized BMDMs (iBMDMs) stably expressing Tet-GSDMD-NT were plated in 96-well plates at 5 × 104 cells per 0.1 ml of complete DMEM. The following day, cells were treated with 1 µg ml−1 doxycycline and 500 ng ml−1 LPS in the presence or absence of 10 µM EGTA-AM. Then 8 h posttreatment, supernatants were removed and 3–5 × 104 of Flt3L-BMDCs were added to each well in 30 µl complete IMDM. After 2 h of incubation, the BMDCs in suspension were transferred to 96-well plates, incubated for an additional 3 h and then analyzed by flow cytometry.
Flow cytometryFor analyzing p-SYK signaling in cDC1 dendritic cells, WT or Clec9a−/− Flt3L-derived BMDCs were washed with PBS and immediately fixed with 1% PFA for 20 min at room temperature. Cells were centrifuged (500g for 5 min) and resuspended in prechilled 300 µl of True-Phos Perm Buffer (cat. no. 425401, Biolegend) and incubated at −20 °C for 45 min. Cells were washed with PBS + 1% FBS and pelleted (2,000g for 5 min). Flt3L-BMDCs were stained for 45 min at 4 °C in PBS with 1% FBS containing the following fluorescently conjugated antibodies and dyes: PE anti-mouse p-SYK (clone l120-722, BD Biosciences, cat. no. 558529), BV711 anti-mouse CD24 (clone M1/69, BD Biosciences, cat. no. 563450), APC anti-mouse XCR1 (clone ZET, Biolegend, cat. no. 148206) and BUV395 anti-mouse CD80 (clone 16-10A1, BD Biosciences, cat. no. 740246). Data were acquired on a BD LSRFortessa X-20 (BD Biosciences) and analyzed using FlowJo v.10 software.
For analyzing cDC1 dendritic cell activation by pyroptotic corpses, Flt3L-BMDCs were washed and stained for 20 min at 4 °C in MACS buffer (PBS with 1% FBS, 2 mM EDTA) containing the following fluorescently conjugated antibodies and dyes: FITC anti-mouse CD11c (clone N418, Biolegend, cat. no. 117306), PerCP-eFluor 710 anti-mouse SIRP alpha (clone P84, Invitrogen, cat. no. 46-1721-82), PE/Cyanine7 anti-mouse CD24 (clone M1/69, Biolegend, cat. no. 101822), PE anti-mouse CD86 (clone GL-1, Biolegend, cat. no. 105008), BV711 anti-mouse CD80 (clone 16-10A1, Biolegend, cat. no. 104743), APC anti-mouse I-A/I-E (MHCII) (clone M5/114.15.2, Biolegend, cat. no. 107614), and Live/Dead Fixable Violet Dead (ThermoFisher, cat. no. L34964). Data were acquired on a FACSymphony A5 SE (BD Biosciences) and analyzed using FlowJo.
In vivo LPS challengeWT or Gsdmd−/− mice (females at 8–10 weeks old, 4–5 mice per group) were injected with 200 µl of 10% thioglycollate broth (108190, Sigma Aldrich) into the peritoneal cavity. Three days later, mice were intraperitoneal challenged with either PBS or with 200 µl of 1 mg ml−1 LPS (L8643, LPS Pseudomonas aeruginosa, Sigma Aldrich). After 4 h, mice were humanely euthanized using CO2. Peritoneal lavage was performed using 5 ml of sterile PBS, and pelleted by centrifugation at 500g for 5 min. Cell pellets were resuspended, counted and equal amounts of cells were centrifuged onto poly-l-lysine-coated (P4707, Sigma Aldrich) coverslips and fixed in 4% PFA for imaging.
Statistical analysisStatistical analyses were performed in GraphPad Prism, and are detailed in each figure legend. Violin plots show quartiles (dotted lines) and mean (solid line). Not significant (NS), P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 and ****P ≤ 0.0001.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)