
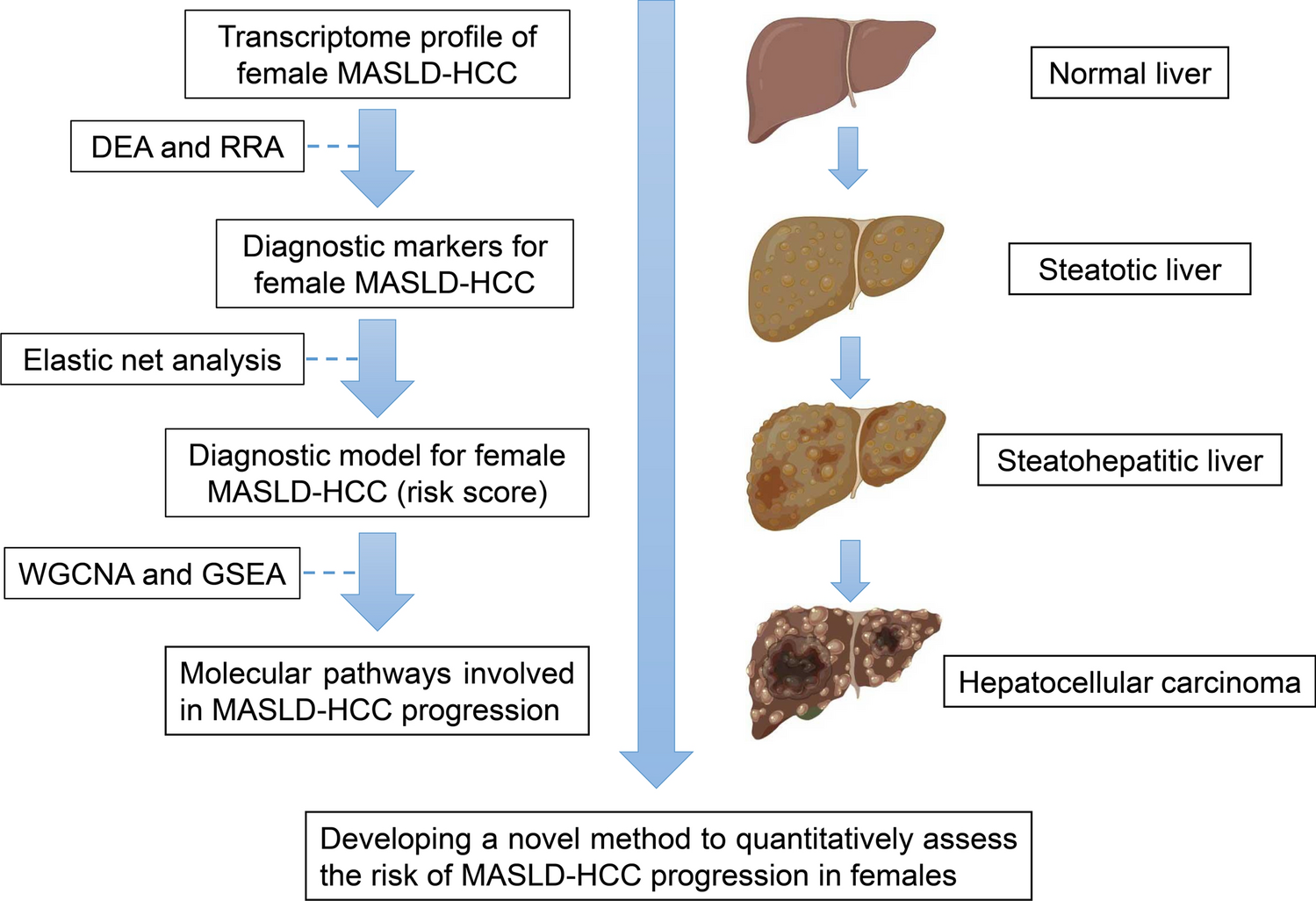
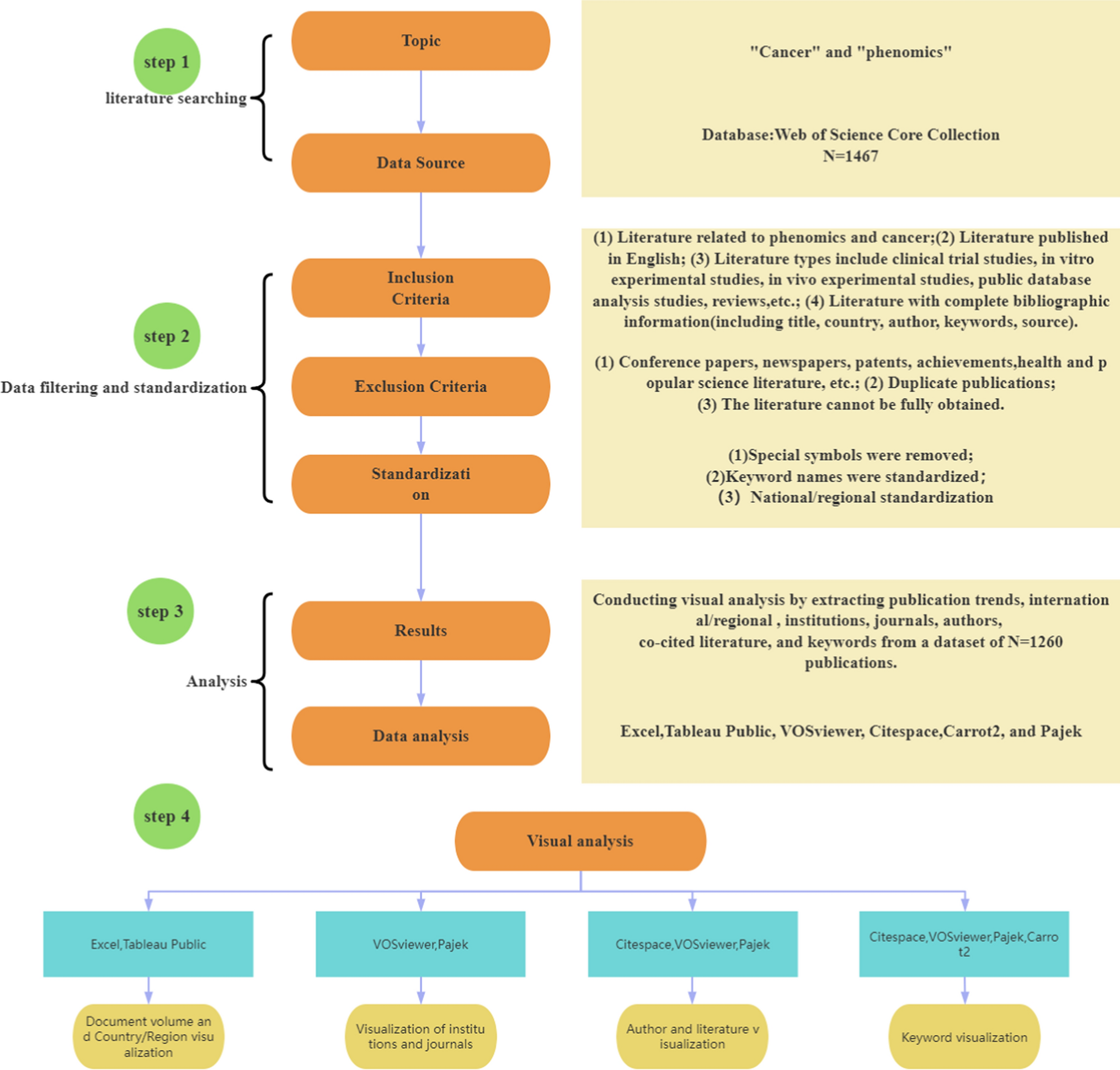
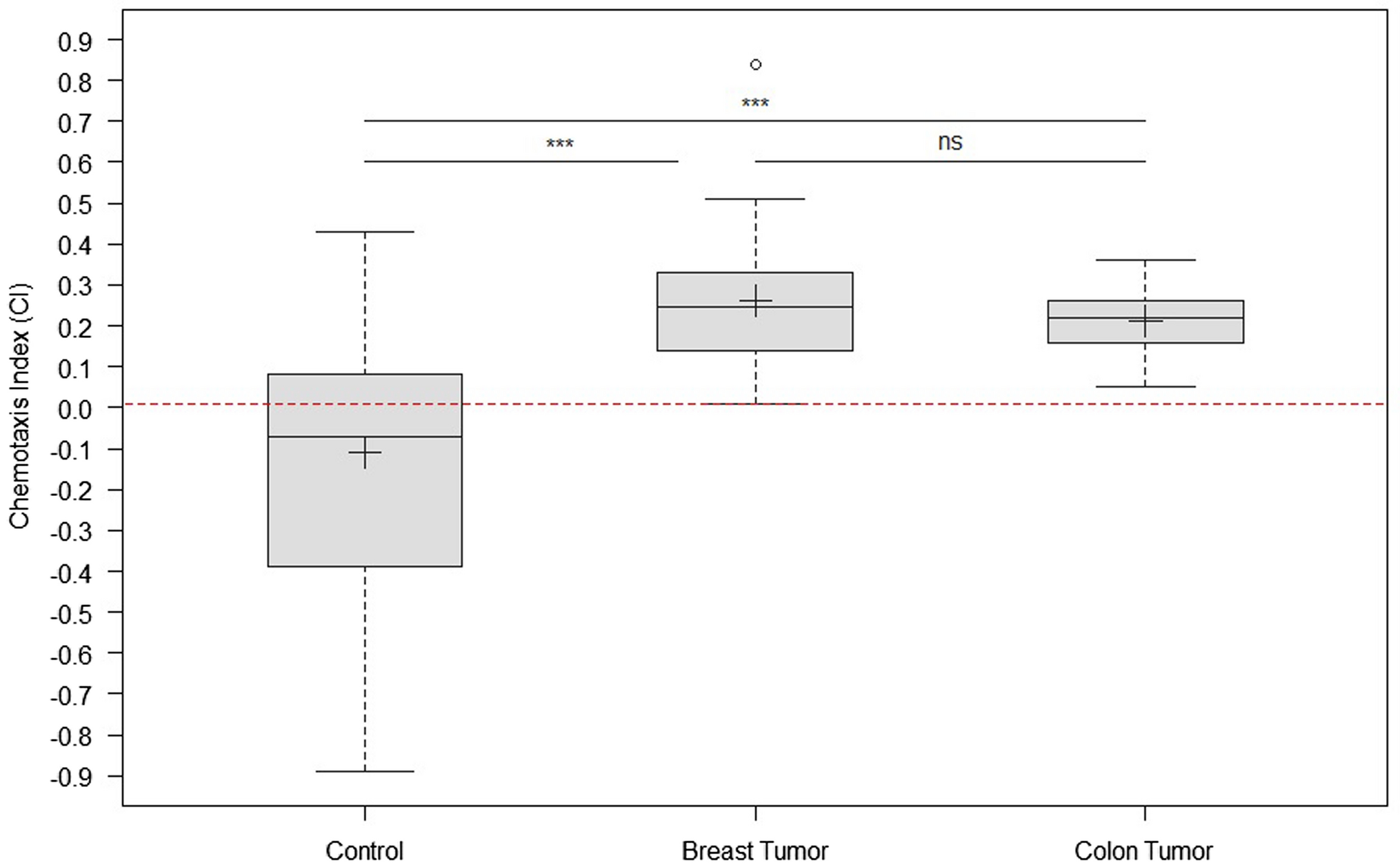
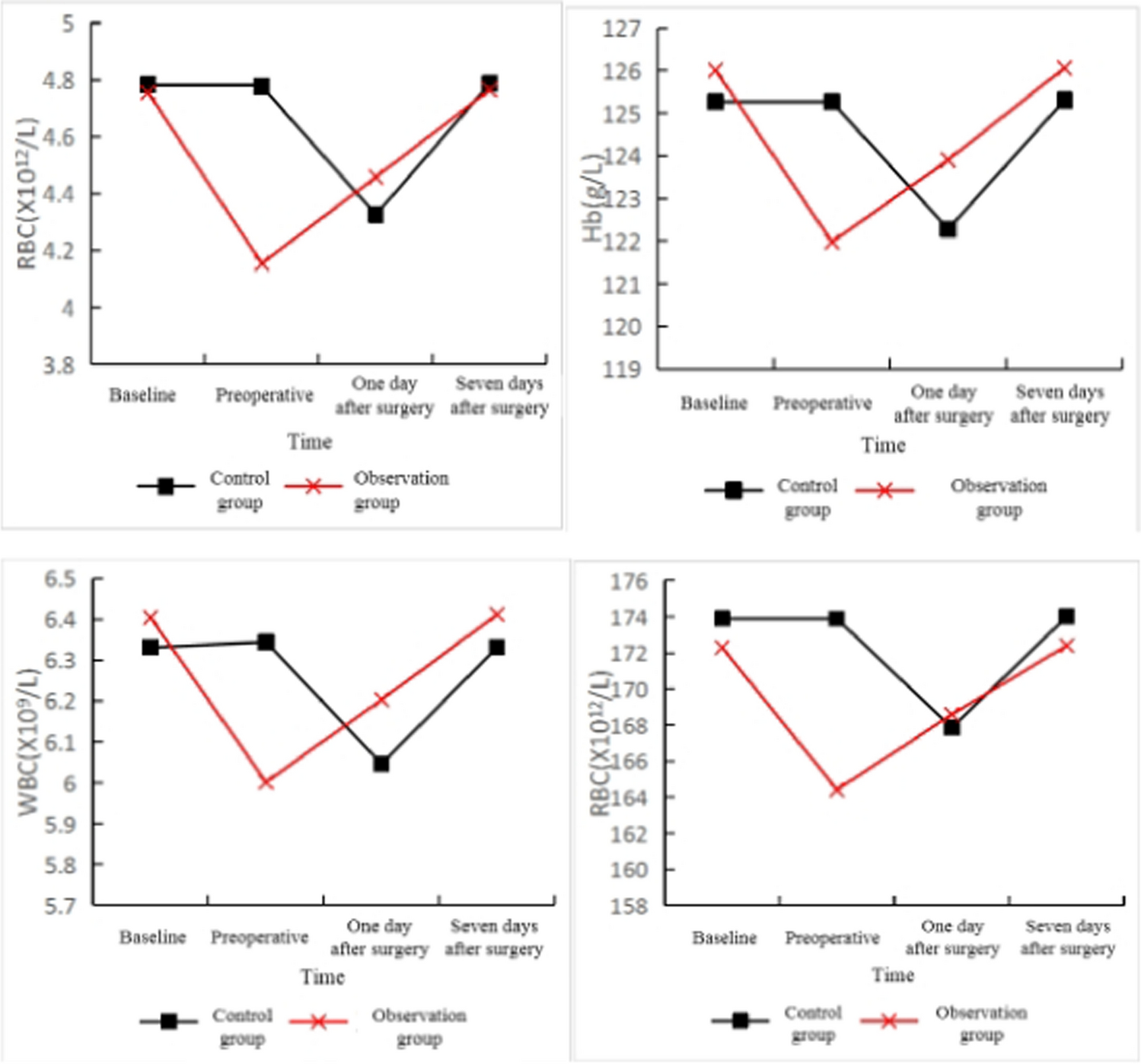
記住我





The differential analysis identified 14 ICD-related genes with significant variations between normal and tumor samples (Fig. 2A), including CD4, CASP8, HSP90AA1, BAX, CALR, and PDIA3, all exhibiting highly significant differences (P-values < 0.001); ENTPD1, TLR4, IL10, and IL17RA showed notable differences (P-values < 0.01); while IL6, IL1B, NLRP3, and HMGB1 also demonstrated differences (P-values < 0.05). The constructed PPI network, depicted in Fig. 2B, is based on these differential ICD-related genes. Each network node symbolizes the proteins produced by a single, protein-coding gene locus, with edges indicating protein-protein interactions. Different colors represent evidence validated from various databases or experimental sources.
Fig. 2
Differential expression of 14 ICD-related genes. A Heat map illustrating the expression levels of 14 ICD-related genes. The horizontal axis represents sample names (blue for normal samples, red for tumor samples), and the vertical axis lists the names of differential ICD related genes. Genes marked with an asterisk indicate significant differences between normal and tumor samples. Significance levels are denoted as follows: ***p-value < 0.001, **p-value < 0.01, *p-value < 0.05. Blue signifies low expression, white indicates intermediate expression, and red denotes high expression. B PPI network among the 14 differential ICD-related genes
3.2 Identification of the ICD subgroupsOur objective was to achieve high correlation within subtypes and low correlation between them. Cluster analysis indicated that the highest consistency within groups occurred when K = 2, leading to the division of Asian HCC patients into two subtypes: C1 and C2 (Fig. 3A). Subsequent analysis of ICD-related gene expression revealed that subtype C1 corresponds to the high ICD expression group, while C2 aligns with the low ICD expression group (Fig. 3B).
Fig. 3
Subgroup classification of Asian HCC samples based on ICD-related genes. A Heat map showing the consensus clustering result (k = 2) for 34 ICD-related genes in 161 Asian HCC samples. B Heat map displaying the expression of 34 ICD-related genes in C1 and C2 subtypes. The horizontal axis represents sample names, with the front green section indicating C1 subtype samples and the rear red section representing C2 subtype samples. The vertical axis displays the expression levels of ICD-related genes. Color coding for expression levels is as follows: blue for low expression, white for intermediate expression, and red for high expression
3.3 Subgroup survival analysisSurvival analysis revealed that the ICD high expression group exhibited a lower survival rate compared to the ICD low expression group, with a statistically significant difference (P-value < 0.05) as shown in Fig. 4.
Fig. 4
Kaplan–Meier (K–M) survival curves for ICD high and low expression groups. This figure presents the survival curves, with the horizontal axis indicating survival time in years and the vertical axis showing the survival rate, which diminishes over time. The blue curve represents the low ICD expression group, while the red curve denotes the high ICD expression group
3.4 Subgroup difference analysisThrough differential analysis, we identified genes with varying expressions between the ICD high expression group and the ICD low expression group, as illustrated in Fig. 5A. GO enrichment analysis revealed that these differential genes were predominantly enriched in functions such as positive regulation of cell activation, external side of plasma membrane, and antigen binding, among others (Fig. 5B). Similarly, KEGG enrichment analysis showed an enrichment of differential genes in pathways like the PI3K–Akt signaling pathway, cytokine–cytokine receptor interaction, and regulation of the actin cytoskeleton, to name a few (Fig. 5C). Further, through GSEA, we explored potential signaling pathways differentiating the ICD high and low expression groups. Figure 5D, E suggest that pathways associated with the ICD high expression group might include cell adhesion molecules (CAMS), cytokine receptor interaction, etc., whereas the ICD low expression group may be linked with pathways such as butanoate metabolism, glycine serine and threonine metabolism, among others.
Fig. 5
Enrichment analysis of differential genes between ICD high and low expression groups. A Heat map illustrating the differential gene expression in ICD high and low expression groups. B Bubble chart of GO enrichment analysis for differential genes. The horizontal axis shows the gene ratio, and the vertical axis lists the GO categories: Biological Process (BP), Cell Component (CC), and Molecular Function (MF). The circle size indicates the number of genes; larger circles represent greater gene enrichment in GO, and a redder hue indicates higher enrichment levels. C Bubble chart of KEGG enrichment analysis for differential genes. The horizontal axis displays the gene ratio, and the vertical axis shows KEGG pathways. As with GO, circle size denotes the number of genes, and a redder color signifies higher enrichment. D GSEA enrichment results for the ICD high expression group. E GSEA results for the ICD low expression group. The horizontal axis represents the sequenced genes, and the vertical axis shows the enrichment score. Different color curves represent distinct pathways
3.5 Subgroup mutation analysisMutation analysis indicated that genes such as TP53, MUC16, OBSCN, CSMD3, ALB, RYR1, XIRP2, and FAT3 exhibited higher mutation frequencies in the ICD high expression group compared to the ICD low expression group. Conversely, genes including CTNNB1, TTN, PCLO, AXIN1, ARID1A, FLG, HMCN1, ABCA13, APOB, DMD, and CACNA1E showed higher mutation frequencies in the ICD low expression group. Notably, RYR2 maintained the same mutation frequency in both subgroups (Fig. 6).
Fig. 6
Waterfall plot of gene mutations in ICD high and low expression groups. This plot displays gene mutations, with the horizontal axis representing samples and the vertical axis denoting genes. Different colors indicate various mutation types, allowing observation of each gene’s mutation frequency
3.6 Differential analysis of tumor microenvironment in subgroupsIn terms of the tumor microenvironment, stromal and immune cells scored higher in the ICD high expression group compared to the ICD low expression group. Consequently, tumor purity was found to be greater in the ICD low expression group. This difference was statistically significant with a P-value less than 0.001 (Fig. 7).
Fig. 7
Violin plot of tumor microenvironment in high and low ICD expression groups. A The stromal cell score; B the immune cell score; C the composite score; D tumor purity. The color blue represents the low ICD expression group, while red signifies the high ICD expression group. The vertical axis displays the tumor microenvironment score. ***Represents p < 0.001
3.7 Differential analysis of immune cells, HLA genes, and immune checkpoint-associated genes in subgroupsThrough differential analysis, it was observed that only neutrophils showed significant variance between the ICD high and low expression groups. Their prevalence was higher in the ICD high expression group, with a P-value less than 0.05, indicating statistical significance (Fig. 8A). The expression levels of certain HLA genes, as well as immune checkpoint-associated genes, were elevated in the ICD high expression group compared to the ICD low expression group. These differences were statistically significant, with p-values less than 0.05, as elaborated in Fig. 8B, C.
Fig. 8
Differential analysis of immune cells, HLA genes, and immune checkpoint-associated genes in ICD high and low expression groups. A Violin plot illustrating the content of immune cells in both ICD high and low expression groups. The horizontal axis labels the immune cell types, while the vertical axis quantifies their content. Blue denotes the ICD low expression group, and red indicates the ICD high expression group. An asterisk (*) signifies a difference in immune cell content between the high and low expression groups. B Box plot depicting HLA gene expression across the two groups. The horizontal axis shows HLA-related genes, and the vertical axis represents gene expression levels. Blue signifies the ICD low expression group, red the ICD high expression group. Significance levels are indicated: ***p value < 0.001, **p value < 0.01, *p value < 0.05. C Box plot of immune checkpoint-associated genes expression in the ICD high and low expression groups. The horizontal axis categorizes the sample groups, and the vertical axis measures gene expression. A p-value < 0.05 indicates significant differences
3.8 Construction of an ICD prognostic modelA 5-gene risk-score prognostic model (Fig. 9) was developed in the training group using Lasso–Cox analysis. The formula for the risk score was: risk score = 0.000521452 × ExpBAX + 0.008997493 × ExpCASP8 + 0.001231 × ExpHMGB1 + 0.001407455 × ExpHSP90AA1 + 0.011885139 × ExpIL6 (Table 2). Subsequent validations of the model in the test group revealed, via K–M survival analysis, that the survival rate was higher in the low-risk group compared to the high-risk group in both training and test groups. The P-values were 0.008 in the training group and 0.023 in the test group, both below 0.05, thus statistically significant (Fig. 10A, B).
Fig. 9
Lasso–Cox analysis results in the training group. This figure displays the point of minimum cross-validation error, marked by a dotted line. The number of genes at this point corresponds to the number used in model construction
Table 2 The 5 ICD related genes involved in the construction of the risk score prognostic model Fig. 10
Validation of the prognostic model. A K–M survival curves for the training group. B K–M survival curves for the testing group. The red curve represents the high-risk group, while the blue curve denotes the low-risk group. In both training and testing groups, the low-risk group exhibited significantly higher survival rates and longer survival times than the high-risk group (p < 0.05), indicating statistical significance. C ROC curve for the training group. D ROC curve for the testing group. The horizontal axis represents the false-positive rate, and the vertical axis the true-positive rate. The area under the curve reflects the model’s accuracy. E Risk plot for the training group. F Risk plot for the testing group. Risk scores increase from left to right, with the median value indicated by a dotted line. The left green line represents low-risk patients, and the right red line indicates high-risk patients. G Survival time and status graph for the training group. H Survival time and status graph for the testing group. Red dots signify deceased patients, and green dots indicate living patients. As risk scores rise, survival time decreases and mortality rates incrementally increase
As depicted by the ROC curves in Fig. 10C, D, the AUC values were 0.783 for the training group and 0.655 for the test group, both exceeding 0.5, suggesting the model’s moderate accuracy. Figure 10E, F illustrate the survival curves for the training and test groups, while Fig. 10G, H display the survival status, indicating a decrease in survival time and an increase in the number of deaths with escalating risk values in both groups.
3.9 Validation of the ICD prognostic modelIntegrated data from both training and test groups, we conducted independent prognostic analysis and risk score-immune cell correlation analysis. The results indicated that the risk score prognostic model, based on ICD-related genes, could serve as an independent prognostic factor for the Asian HCC population (Fig. 11A, B). The risk score was found to be negatively correlated with M1 cells (P = 0.033) and γδ T cells (P = 0.024), both statistically significant correlations (Fig. 12).
Fig. 11
Independent prognostic analysis. A Univariate cox independent prognostic analysis. B Multivariate cox independent prognostic analysis
Fig. 12
The p-values for stage and risk score were below 0.05 in both univariate and multivariate Cox independent prognostic analyses, suggesting their validity as independent prognostic factors.
3.10 Clinical correlation analysisWe found that risk scores were related to age and pathological grade (P = 0.009, 0.010, respectively). Specifically, risk scores were higher in the ≤ 65y group than in the > 65y group (Fig. 13A). And compared to the grade 1–2 group, risk scores were lower in the grade 3–4 group (Fig. 13B). Although there was a tendency to show higher risk scores in the stage III–IV group than in the stage I–II group, this was not statistically significant (P = 0.057, Fig. 13C).
Fig. 13
Clinical correlation. A age; B grade; C stage
留言 (0)