
記住我


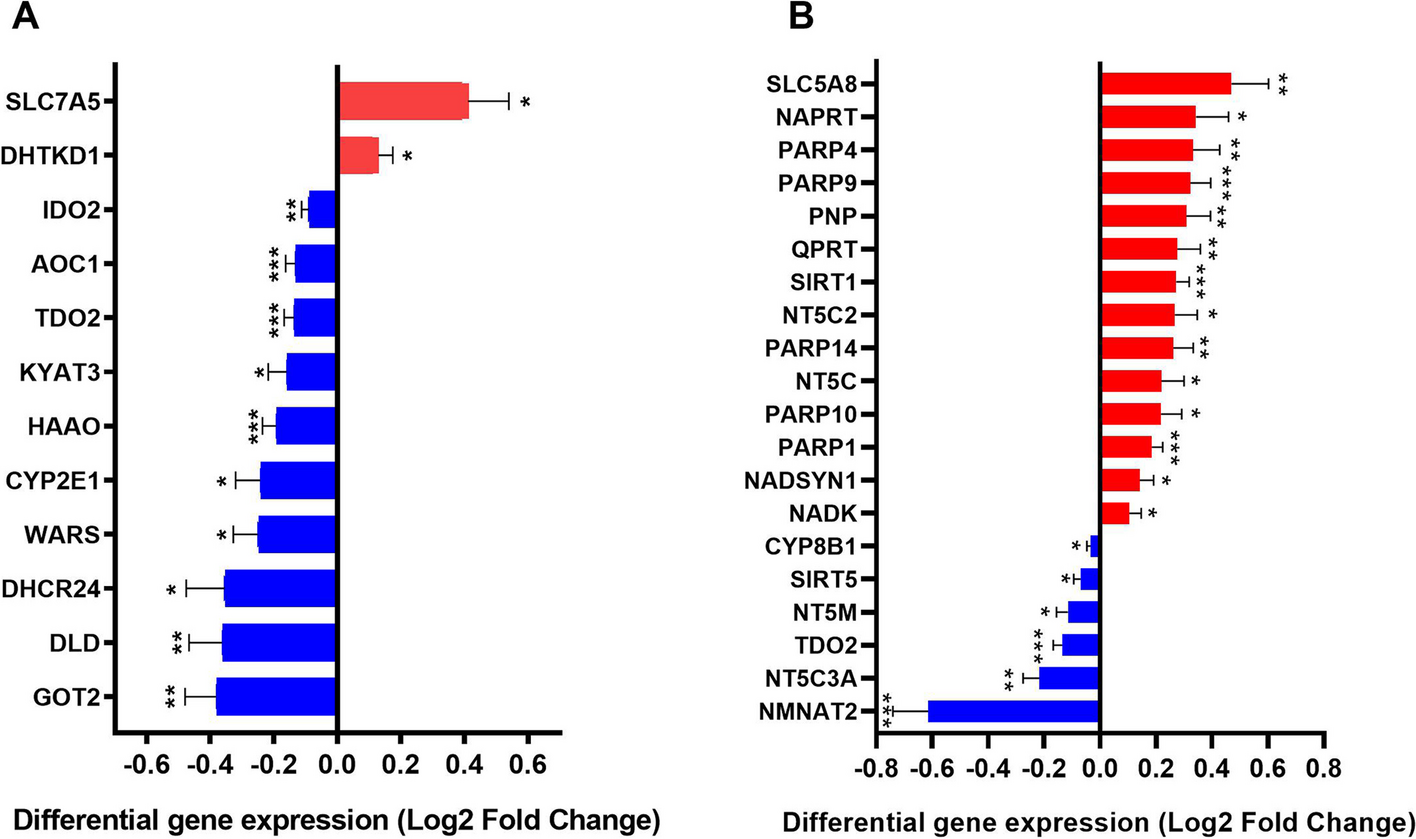


Based on the systematic research of three databases, it resulted in a gene set consisting of 60 TRP- and 57 NAD-associated genes (Tables S1 and S2). Out of the 60 identified TRP pathway-associated genes, 56 genes were included in the microarray analysis. After FDR correction, 12 of these genes (2 up-regulated [SLC7A5, DHTKD1] and 10 down-regulated [TDO2, HAAO, AOC1, GOT2, IDO2, DLD, SLC7A5, WARS, CYP2E1, DHCR24, KYAT3]) showed significant differential mRNA expression in patients with AD versus controls (Fig. 1A; Table S4). In addition, microarray data were available for 52 of the 57 NAD pathway-associated genes. After FDR correction, mRNA levels of 20 genes showed statistically significant differences with 6 genes showing lower expression (CYP8B1, SIRT5, NT5M, TDO2, NT5C3A, NMNAT2) and 14 genes showing higher expression (SLC5A8, NAPRT, PARP4, PARP9, PNP, SIRT1, QPRT, PARP14, NT5C2, PARP10, NT5C, PARP1, NADSYN1, NADK) (Fig. 1B; Table S5).
Fig. 1
TRP and NAD pathway associated genes show significant differential gene expression. Shown are all the genes in the TRP and NAD pathway with up (red) or down (blue) regulation in the middle temporal gyrus of AD patients compared to age-matched controls. Differential expression is presented as log2 fold change. Differential regulation was assessed using false discovery rate (FDR) test and q < 0.05 was considered significant. * q < 0.05, ** q < 0.01 and *** q < 0.001. Error bars represent standard error of mean (SEM)
Transcriptomic enrichment analysisTo examine whether expression changes in TRP- and NAD-related genes are overrepresented in AD, an enrichment analysis was conducted based on the transcriptomic profiling within the MTG. It identified 31,726 genes of which 11,459 genes were identified to be nominally (limma p-value < 0.05) differentially expressed between AD patients and age-matched control individuals [15]. Once adjusting for multiple testing (limma FDR q-value < 0.05), 7,776 genes were significantly differentially expressed at the mRNA level, resulting in an overall enrichment score of 24.5% (7,776/31,726 genes) as a baseline. The overall TRP pathway only displayed a 21.4% enrichment score (12/56 genes), indicating this gene set overall did not display a significant enrichment in view of differentially expressed genes (Table S4). However, the enrichment score for the KP, which accounts for > 90% of all TRP metabolism, showed a 41.7% enrichment score (5/12 genes) indicating a significant enrichment in differentially expressed genes within the KP-associated gene set (Table S4). Lastly, the NAD pathway showed a 38.5% enrichment score (20/52 genes) indicating a significant enrichment in differentially expressed genes within the NAD-associated gene set (Table S5). NAD pathway is divided into two sub-pathways, Salvage pathway and Preiss-Handler pathway, and both pathways were enriched (Salvage: 34.14% [14/41 genes]; Preiss-Handler: 50% [8/16 genes]).
Correlation between gene expression and AD pathologiesCorrelation analysis was conducted to assess the association between mRNA expression and AD pathologies (total Aβ plaque and tau tangle). Based on the KP-associated genes with multiple test correction, TDO2, GOT2, and IDO2 showed significant negative correlation with both plaque and tangle, while KYAT3 only showed negative correlation with total plaque (Table S6). In a similar manner, the NAD-associated genes, SIRT1, PARP9, PARP14, QPRT, PARP10, NAPRT, NADSYN1, and NADK showed positive correlation in both plaque and tangle, while PNP, NT5C2, PARP1, and PARP4 was positively correlated in plaque only. Additionally, NMNAT2 and NT5C3A showed negative correlation in both plaque and tangle (Table S6). Based on these results, several significantly differentiated expressed genes in both KP- and NAD-associated pathways were significantly correlated in AD pathologies even after multiple test corrections.
Alterations of DNA (hydroxy)methylation in the brain of patients with ADNext, we investigated whether the AD-specific TRP- and NAD-associated mRNA profiles were associated with DNA methylation differences at the level of 5mC, 5hmC, or 5uC. Within the TRP pathway, differences in 5mC, 5hmC, and 5uC levels showed nominal significance (p-value < 0.05) for 19/847 probes, 18/510 probes, and 29/847 probes, respectively (Fig. 2A-C; Tables S7-S9). Within the NAD pathway, 5mC, 5hmC, and 5uC showed nominally significant differences for 20/1009 probes, 18/632 probes, and 35/1009 probes, respectively (Fig. 2D-F; Tables S10-S12). These differentially (hydroxy)methylated probes were associated with 18 significantly differentially expressed genes. Of these, five genes exhibited significant differences in various forms of methylation (i.e. 5mC, 5hmC, and/or 5uC) at the same CpG site. An overview of the differently expressed genes and DNA (hydroxy)methylation levels within the TRP metabolic pathway and NAD pathway are shown in Fig. 3. It is worth noting that, in the DNA (hydroxy)methylomic profile analyses, the TRP-associated genes ASMT and KYAT1 were not included in this analysis, as data on DNA (hydroxy)methylomic- profiles were not available for these genes. In addition, ALDH9A1 and SLC36A4 were excluded from the 5hmC analysis due to missing data. Concerning the NAD pathway, the NADK2, NMNAT1, and NMRK genes were not included in the analysis, as data were not available for these genes. In addition, PARP2 was excluded from the 5hmC analysis due to missing data.
Fig. 2
TRP and NAD pathway associated genes show significant differential DNA (hydroxy)methylation levels. Shown are significant CpG sites with its corresponding gene name in the tryptophan metabolic pathway (A-C) and NAD pathway (D-F) with up (red) or down (blue) regulation in the middle temporal gyrus of AD patients compared to age-matched controls. A, D Differential methylation (5mC) expression. (B, E) Differential hydroxymethylation (5hmC) expression. C, F Differential unmodified (5uC) expression. Differential expression is presented as log2 fold change. Differential regulation was assessed using limma differential DNA (hydroxy)methylation level analysis and p < 0.05 was considered significant. * p < 0.05 and ** p < 0.01. Error bars represent standard error of mean (SEM)
Fig. 3
Overview of the differentially expressed genes and DNA (hydroxy)methylation levels within the TRP metabolic pathway and NAD pathway. Within the TRP metabolic pathway, a total of 12 genes exhibited significant differential gene expression, while 20 genes were differentially expressed within the NAD pathway. Genes exhibiting up-regulation in expression are highlighted with red box, whereas those with down-regulation are indicated by blue box. Genes with altered DNA (hydroxy)methylation levels are denoted by circular markers, with distinct colors representing specific modifications: 5mC (green), 5hmC (purple), and 5uC (orange)
Correlation between gene expression and DNA (hydroxy)methylated probesThe differentially (hydroxy)methylated probes were linked to several significantly differentially expressed genes. Seven of these genes showed significant differences in different types of methylation modifications for the same CpG site. Moreover, two differentially hydroxymethylated CpG sites within the SLC7A5 gene, i.e., cg10169763 and cg09409405, displayed a negative correlation between 5hmC levels and mRNA expression (Fig. 4A, B). For 5uC, both cg19571004 (CYP2E1) and cg01812894 (ALDH1A1) showed a positive correlation with mRNA expression (Fig. 4C, D). In relations to the NAD pathway, nine CpG sites displayed significant correlations between DNA (hydroxy)methylation and mRNA expression (Fig. 5). Concerning 5mC, three CpG sites (cg21580588 [NADK]; cg09185911 [NADK]; cg14750551 [PARP14)]) showed a positive correlation (Fig. 5A, B and F), while two other CpG sites (cg10582690 [SIRT1] and cg11229284 [PARP14]) showed a negative correlation (Fig. 5C, D). At the 5hmC level, again, cg14750551 (PARP14) showed a significant negative correlation (Fig. 5F), while cg24937136 (PARP1) was positively correlated (Fig. 5E) to its mRNA expression profile. Finally, for 5uC, cg16373880 (NMNAT2) showed a positive correlation (Fig. 5G), whereas two CpG sites (cg05215649 [NADSYN1] and cg15824543 [NADK]) showed a negative correlation (Fig. 5H, I) with the corresponding mRNA levels.
Fig. 4
Spearman’s correlation analysis of TRP pathway. Spearman’s correlation analysis between methylation (■5hmC and ▲5uC) and mRNA expression levels. Spearman’s correlation analysis are presented with Spearman’s R-value, 95% confidence interval (CI), and p-value
Fig. 5
Spearman’s correlation analysis of NAD pathway. Spearman’s correlation analysis between methylation (●5mC, ■5hmC, and ▲5uC) and mRNA expression levels. Spearman’s correlation analysis are presented with spearman R-value, 95% confidence interval and, p-value
Gene regulatory network and network perturbation analysisA GRN was generated through MetaCore (Clarivate Analytics), which builds gene networks by implementing direct functional interactions between genes acquired from experiments-based literature reports. In combination with identifying a GRN, we identified network perturbation candidates that have the potential to induce a positive phenotypic transition, i.e., as in moving from a diseased to a healthy GRN (Table 4). Concerning the TRP pathway, the reconstructed control network comprised 22 nodes and 30 interactions (Fig. 6A), while the AD network comprised of 22 nodes and 25 interactions (Fig. 6B). In the associated perturbation analysis, 9 genes were identified whose alteration holds the potential to revert the gene expression program from a diseased towards a healthy state. Two of these genes (IDO2 and CYP2E1) showed significant changes in differential mRNA expression after FDR correction when comparing AD cases with control subjects. The highest perturbation scores were obtained for a 3-gene perturbation combination, involving CAT, IDO2, and CYP2E1 (Table 4).
Table 4 Perturbation scores of TRP- and NAD- pathway associated genes Fig. 6
Gene regulatory network (GRN) of TRP and NAD pathway. A TRP GRN representing the control phenotype and containing 22 nodes and 30 interactions, (B) TRP GRN representing Alzheimer’s disease (AD) phenotype and containing 22 nodes and 25 interactions, (C) NAD GRN representing the control phenotype and containing 13 nodes and 21 interactions, (D) NAD GRN representing Alzheimer’s disease (AD) phenotype and containing 14 nodes and 24 interactions. Green line indicates gene activation while red line indicates gene inhibition
Similarly, GRNs for the NAD pathway were constructed. The control NAD network comprised 13 nodes and 21 interactions (Fig. 6C), while the AD network comprised 14 nodes and 24 interactions (Fig. 6D). Furthermore, in the perturbation analysis, 8 genes were identified, of which 2 genes (SIRT1 and PARP1) showed significant differential mRNA expression after FDR correction. The highest perturbation score was obtained for a 2-gene perturbation combination, involving SIRT1 and PARP1 (Table 4).
IDO2 methylation in AgeCoDeBased on the results from the molecular profiles, correlations, and computational models, IDO2, SLC7A5, and PARP14 were selected as potential candidate genes for further investigation. As such, an independent longitudinal cohort (AgeCoDe), with DNA derived from blood available, was used in an attempt to validate the MTG methylation findings. In the AgeCoDe cohort, 8 out of 106 CpG sites across these genes showed nominal significant differential DNA methylation (Table S13), of which only cg11251498 (IDO2) was shown nominal significance for both MTG and AgeCoDe while the rest were not found in the MTG analysis. Further analysis, after adjusting for age and gender at both time points, revealed that the cg11251498 (IDO2) methylation level was significantly higher in AD converters compared to controls at baseline (p = 0.001). Although methylation levels were elevated in AD patients after a 4.5-year follow-up, this difference was not statistically significant (p = 0.051) (Table 2).
IDO2 pyrosequencing in BBACLIn order to validate cg11251498 (IDO2), we pyrosequenced this locus in DNA from blood samples of subjects from an independent longitudinal cohort (BBACL). At baseline, after correcting for covariates, cg11251498 (IDO2) did not show a significant difference in DNA methylation levels when comparing SCD, MCI, and dementia patients (model 1, p = 0.58; model 2, p = 0.55). Similarly, no difference in IDO2 DNA methylation was observed when comparing future converters and non-converters (MCI-D, MCI-MCI) at baseline (model 1, p = 0.38; model 2, p = 0.32) (Table 3). Interestingly, a strong negative association between IDO2 DNA methylation and age was seen both when comparing SCD, MCI, and dementia patients (model 1: β = −0.11, S.E. = 0.04, p = 0.006, 95% CI = −0.19 to −0.031) and when comparing converters to non-converters (model 1: β = −0.14, S.E. = 0.053, p = 0.009, 95% CI = −0.24 to −0.035). Moreover, an age association was still significant for both comparisons when applying model 2 (Table 5). Age significance was neither observed in AgeCoDe nor in the MTG data (Table S14). Finally, cg11251498 (IDO2) showed no significant association with cognition when comparing SCD, MCI, and dementia patients at baseline (model 1, p = 0.60; model 2, p = 0.15) and when comparing future converters and non-converters (model 1, p = 0.81; model 2, p = 0.30) (Table 5).
Table 5 IDO2 methylation association with age and cognitive test in the BBACL cohort
留言 (0)