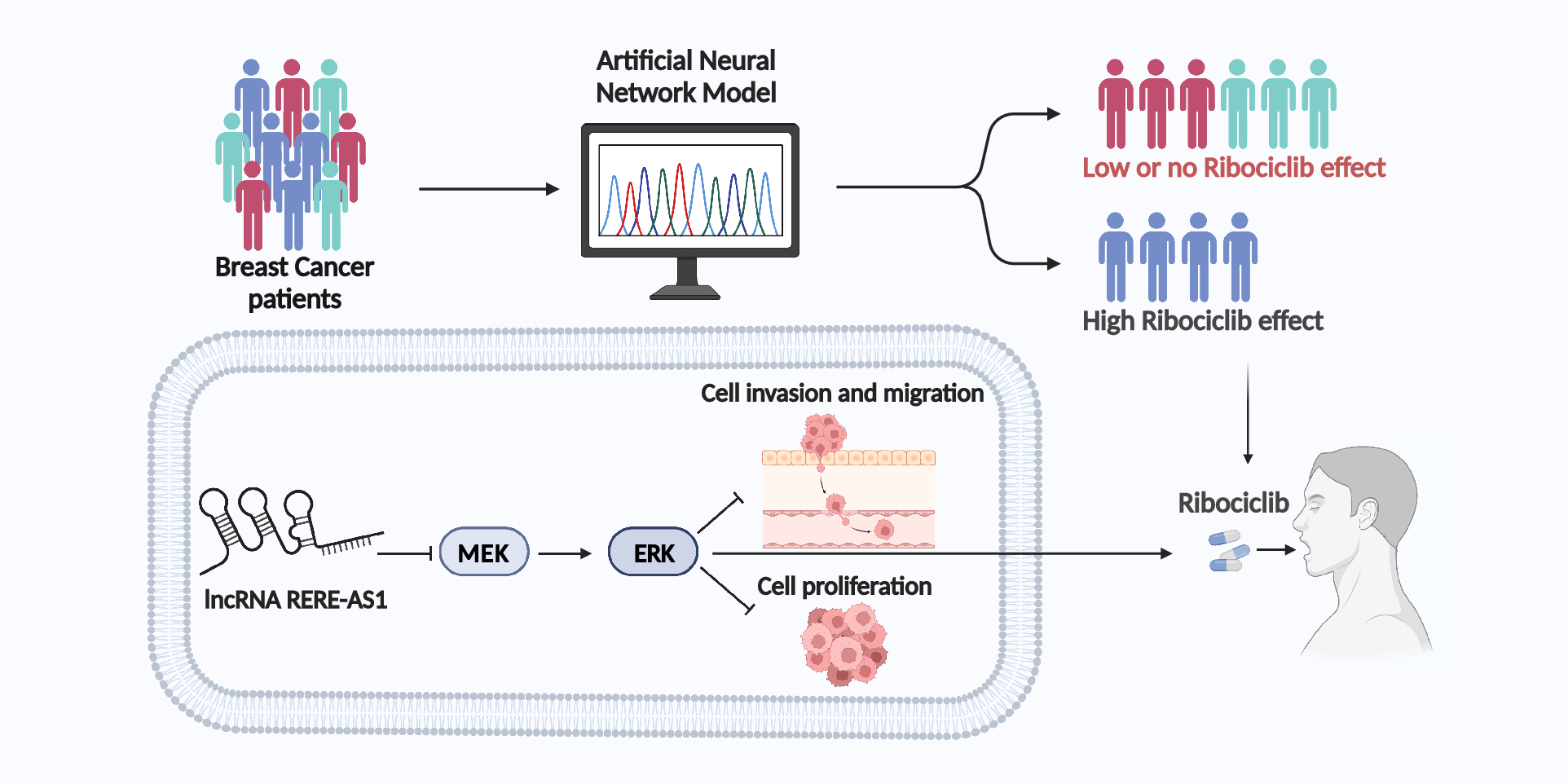
Prediction of half-maximal inhibitory concentration (IC50) values for ribociclib
We obtained transcriptome data and clinicopathological factors and survival data of patients with cancers through the Cancer Genome Atlas (TCGA) database. The raw data was then processed by converting the counts into transcripts per million and normalizing these values using the formula log2 (TPM + 1).
Utilizing the normalized transcriptome data of TCGA-BRCA along with the Genomics of Drug Sensitivity in Cancer v2 (GDSC2) database, we determined the IC50 value of ribociclib for each BC patient by employing the “oncoPredict” R package. The “oncoPredict” R package uses in vitro gene expression and drug sensitivity data to train models, which are then used to predict drug sensitivity in new gene expression datasets [12]. Using the R package “survminer”, we identified the optimal cutoff value for classifying patients into groups of drug sensitivity and insensitivity.
Screening of lncRNAs related to ribociclib
We utilized Spearman correlation analysis and the “limma” R package to identify lncRNAs associated with ribociclib. Differential analysis and prognostic analysis were further used to screen lncRNA. The random forest and lasso algorithms were employed utilizing the “randomForest” and “glmnet” R packages, respectively, to identify the lncRNAs that are most critical for classification or prediction. A Venn diagram was created using the “ggplot2” and “VennDiagram” R packages to display the overlap of lncRNAs that were identified using both random forest and lasso algorithms. The receiver operating characteristic (ROC) curve and area under the curve (AUC) value for the lncRNA data was conducted using the “pROC” package.
Construction and validation of artificial neural network model (ANNM)
Patients were randomly divided into training and validation sets at a 7:3 ratio using the “createDataPartition” function from the “caret” package. The “neuralnet” R package was utilized to build ANNM. The “NeuralNetTools” R package facilitated the visualization and evaluation of the constructed models, providing insights into their structure and performance. The accuracy of ANNM was assessed by employing ROC curve analysis, facilitated by the “pROC” package.
Correlation analysis of RERE-AS1 with signatures of cancer hallmarks and clinical pathological characteristics
Cancer hallmark-related gene sets were compiled and analyzed using the “GSVA” package in R, with the “ssgsea” method set as the parameter [13]. Ultimately, the relationship between RERE-AS1 and the scores for cancer hallmarks was evaluated through Spearman correlation analysis.
We used the R package “pheatmap” to generate a heatmap illustrating the differences of clinical pathological characteristics between the high and low expression levels of RERE-AS1. In addition, we analyzed the discrepancies in RERE-AS1 expression between different T and pathological stages with the “stats” and “car” R package. Then, we visualized the results with box plots using the “ggplot2” package. To further explore clinical application of RERE-AS1, we employed the “survival” and “rms” packages to construct nomograms and to plot calibration curves. The “ggDCA” package was used to perform decision curve analysis (DCA).
Construction of RERE-AS1-mediated ceRNA network
We obtained data on the interaction between lncRNAs and miRNAs from miRcode (http://www.mircode.org/index.php), targetscan (http://www.targetscan.org/vert_80/), and miRanda (www.microRNA.org) databases. Additionally, data on the interaction between miRNAs and mRNAs were acquired from miRWalk (http://129.206.7.150/) and the Encyclopedia of RNA Interactomes (http://starbase.sysu.edu.cn/index.php) databases. Finally, based on the results derived from these databases, we visualized the ceRNA interaction network using the software of CytoScape (version 3.9.1).
Functional enrichment analysis
We performed Gene Ontology (GO) functional enrichment analysis and GO classification annotation for mRNA in the ceRNA network using the GO database (https://www.geneontology.org/), and “clusterProfiler” R package. This database was utilized to identify the biological functions represented by enriched GO terms. Additionally, we utilized the “clusterProfiler” R package to conduct Gene Set Enrichment Analysis (GSEA).
Analysis of differentially expressed genes
We used transcriptome sequencing data of RERE-AS1 overexpressing cell lines and control cell lines to obtain differentially expressed genes using the “limma” R package, and created a heatmap to visualize these genes employing the “pheatmap” package. Additionally, we utilized the “ggplot2” package to construct a volcano plot illustrating the upregulation and downregulation of differential genes.
Pan-cancer analysis of RERE-AS1
We used differential analysis to investigate the difference in expression levels of RERE-AS1 between normal and cancerous tissues. The “survival” package was employed to assess the prognostic role, employing both univariate Cox regression analysis and log-rank analysis based on outcomes including overall survival (OS), recurrence-free survival (RFS), disease-specific survival (DSS), and PFS. The diagnostic value across different cancers was evaluated using ROC analysis.
Cell lines, cell culture, and transfection
Human breast cancer cell lines (T47D, MCF-7, BT474, SUM159PT, MDA-MB-231, MDA-MB-453, MDA-MB-468, BT-549, and Hs578T) along with the HEK293T cell line were acquired from Pricella Life Science & Technology Co., Ltd (China). Cell lines such as MCF-7, MDA-MB-231, MDA-MB-453, MDA-MB-468, Hs578T, and HEK293T were propagated in Dulbecco’s Modified Eagle Medium (DMEM, Biological Industries), whereas T47D, BT474, SUM159PT, and BT-549 were grown in Roswell Park Memorial Institute-1640 (RPMI-1640, Biological Industries) medium. Each medium was supplemented with additional fetal bovine serum (Biological Industries) and penicillin/streptomycin (Solarbio, China).
Human OE-RERE-AS1 lentivirus was obtained from GeneChem (China). MCF-7 cells underwent lentiviral infection at a multiplicity of infection (MOI) of 20, and MDA-MB-231 cells were infected at an MOI of 10. Lentivirus containing vector was used as a negative control for all cell infections. Transfection was carried out using HiTransG P transfection reagent (GeneChem, China), followed by treatment with puromycin-containing medium (MedChemExpress, USA) for one week to establish stable RERE-AS1 overexpressing BC cells.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
RNA was isolated using TRIzol reagent (TAKARA, Japan) and subsequently converted to cDNA with a reverse transcription kit from TAKARA (Japan). mRNA levels were quantified using the 2−∆∆Ct method, normalized against the housekeeping gene GAPDH. The sequences of the PCR primers are listed in Additional file1: Table S1. Additionally, nuclear and cytoplasmic fractions were separated using the Cytoplasmic and Nuclear RNA Purification Kit from Norgen Biotek Corp (Canada).
Tissue specimens
The frozen tissues from 20 cases of tumor specimens and 13 cases of the non-tumoral surrounding tissue specimens were used for RT-qPCR assay. All samples were stored at − 80 °C from the time of harvest. The study was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (bc20240081) and was conducted in accordance with the Declaration of Helsinki.
Cell viability assay
Cells were plated in 96-well plates at a density of 1000 cells per well. Under light-protected conditions, 10 μL of Cell Counting Kit-8 (CCK-8) reagent (Solarbio, China) was added to each well, followed by light-protected incubation in a cell culture incubator for 2 h. cell viability was measured by reading the absorbance at 450 nm using a microplate reader (Bio-Rad, USA). The concentration of ribociclib necessary to achieve a 50% inhibition of cell growth was determined using viability curves obtained from the CCK-8 assay.
Colony formation assay
A total of 1 × 103 cells were placed into each well of 6-well plates and grown for a period of 7 to 21 days. After visible clone formation, the culture medium was discarded, and the cells were fixed with 4% paraformaldehyde for 15 min, stained with 1% crystal violet for 10 min, and then counted. These experiments were conducted in triplicate, with statistical significance determined by the Student’s t-test.
5-Ethynyl-2′-deoxyuridine (EdU) incorporation assay
15 × 104 cells were inoculated into a 12-well plate and cultured overnight. Cells were added with 50 µM EdU reagent (Beyotime, China), incubated in the incubator for 2 h, fixed with 4% paraformaldehyde, and stained with 594 azide. The nucleic acid was stained with Hoechst 33342. Capture images using an inverted fluorescence microscope (Olympus) and analyze staining results using ImageJ software.
Transwell assay
Migration and invasion experiments were conducted using 24-well plates with chambers (Corning, USA) with a pore size of 8 μm. In the invasion assay, the upper chamber was added with 200 μL of MCF-7 at a concentration of 10 × 105 cells/mL and 200 μL of MDA-MB-231 at a concentration of 5 × 105 cells/mL. In the migration assay, the upper chamber was loaded with 200 μL of MCF-7 at a concentration of 5 × 105 cells/mL and 200 μL of MDA-MB-231 at a concentration of 2.5 × 105 cells/mL. The lower chamber was filled with 600 μL of medium containing 20% serum. MCF-7 were cultured for 72 h and MDA-MB-231 cells for 12 h [14, 15]. Cells that migrated through the chambers were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Ten random fields were captured using an inverted optical microscope (Olympus), and cell numbers were quantified using ImageJ software.
Scratch test
Approximately 100 × 104 cells were seeded into each well of a 6-well plate and cultured overnight. Using a 20 µl pipette tip, scratches were made vertically across the confluent cell layer at the bottom of the culture dish. After washing with PBS, images were captured under a microscope. The scratches were photographed again at 24 and 48 h after cultivation. The migration area of the cells was measured using both Adobe Photoshop and ImageJ software.
Western blot
Cell and tissue proteins were extracted using pre-prepared RIPA buffer (Beyotime, Shanghai, China) supplemented with proteinase and phosphatase inhibitors. Following protein denaturation, the cell lysates were subjected to electrophoresis on polyacrylamide gels (Epizyme, Shanghai, China). After electrophoresis, proteins were transferred onto a nitrocellulose membrane (Millipore, Germany). The membrane was blocked with 5% bovine serum albumin, Tris-buffered saline, and 0.2% Tween, and then incubated overnight at 4 °C with primary antibodies. This was followed by incubation with horseradish peroxidase-conjugated secondary antibodies (ZSGB-BIO, #ZB-2305 and ZB-2306) for 45 min at room temperature. The bands were visualized using the Immobilon Western HRP Substrate (Millipore, Germany) and detected with an ImageQuant LAS4000 system (GE Healthcare Life Sciences). Details of the antibodies used are listed in Additional file1: Table S2.
Subcutaneous xenograft models
To establish an ex vivo xenograft tumor model, we selected 16 NOD/ShiLtJGpt mice (NCG mice) from GemPharmatech Co., Ltd. (China) and divided them into two groups (control group and overexpression group). MDA-MB-231 cells were orthotopically inoculated into the abdominal mammary fat pad of 6-week-old female NCG mice, with each mouse receiving an injection of 5 × 106 cells in suspension. Tumor volume was assessed weekly using digital calipers. At the conclusion of the animal experiments, euthanasia was performed on the mice, and tumor tissues were excised for weighing and photography. The mouse experiments were conducted in accordance with the protocol approved by The Animal Ethical and Welfare Committee of Tianjin Medical University Cancer Institute & Hospital.
Statistical analysis
Statistical analyses were conducted using Prism 9.5.1 and R software 4.2.2. The Spearman’s correlation test was employed to assess the relationships between numerical variables. Pairwise comparisons were made using Student’s t-test. One-way analysis of variance (ANOVA) was utilized to identify differences among groups for each assay, with Tukey’s post hoc test applied to determine pairwise differences. p values less than 0.05 were considered significant.








留言 (0)