
記住我

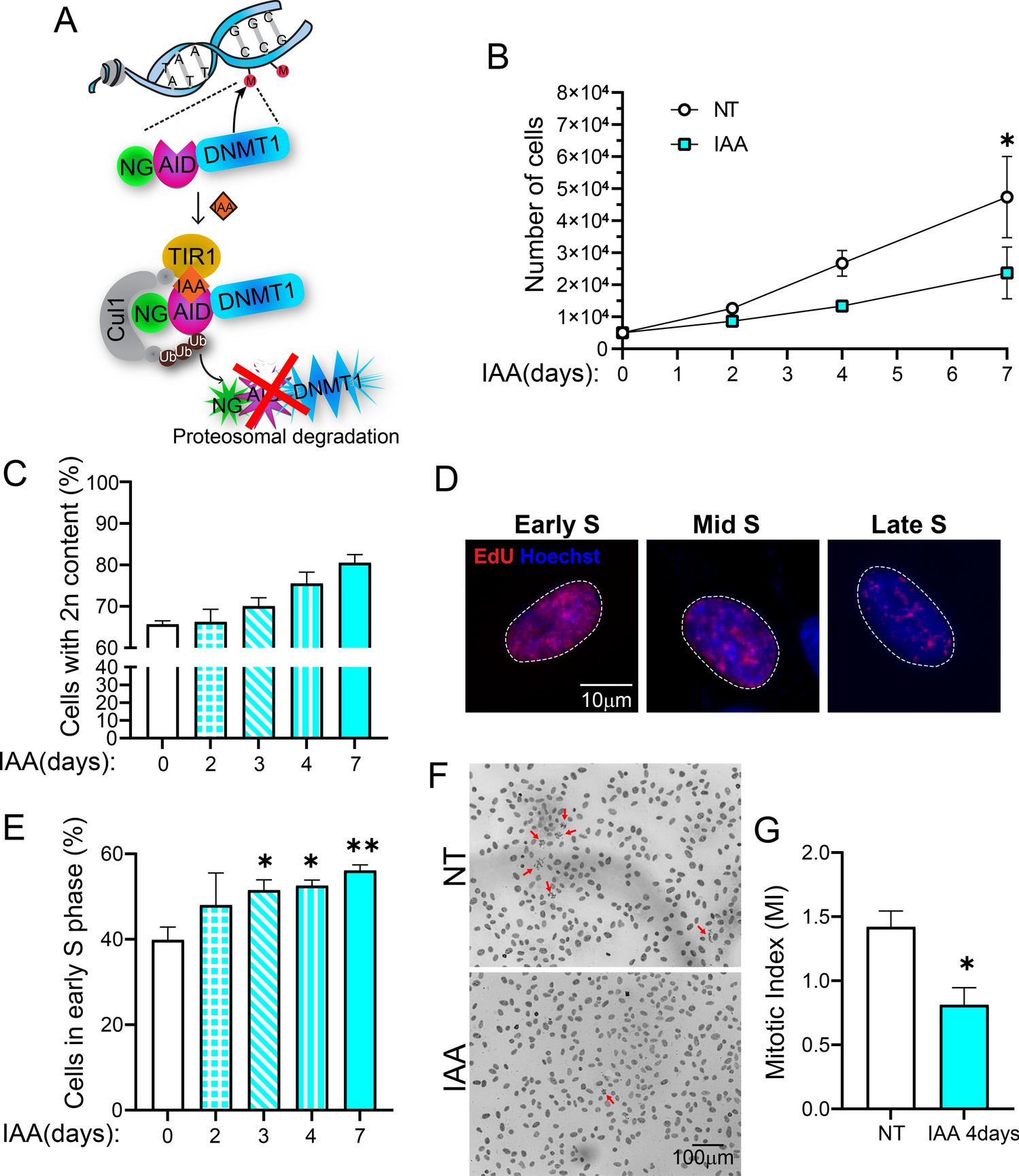






We have previously shown that in HEK293 cells, lack of ESCRT-I causes a very prominent activation of inflammatory NFκB-dependent signaling [30] and lysosomal starvation-related TFEB/TFE3-dependent signaling [14]. However, whether removing ESCRT-I alters the expression of metabolic genes in these cells has not been investigated. Hence, we performed a microarray analysis of HEK293 cells in which we depleted the ESCRT-I components, TSG101 or VPS28 proteins using two distinct siRNAs (designated #1 and #2) against each component (Fig. 1A) and analyzed the cells three days post transfection (3 dpt). Depletion of one of these proteins led to removal of the other one (Fig. 1A), consistent with destabilization of the whole complex that occurs upon depletion of any of its core components [31, 40]. Both siRNAs targeting VPS28 were similarly efficient in reducing the levels of VPS28 and TSG101 proteins, whereas siTSG101#1 was less efficient than siTSG101#2 in depleting TSG101 and removing VPS28 (Fig. 1A).
Fig. 1
ESCRT-I dysfunction leads to reduced expression of genes involved in oxidative metabolism of amino acids and fatty acids. A Western blots showing the depletion efficiencies of ESCRT-I subunits, TSG101 or VPS28 using two single siRNAs for each component (siTSG101#1 or siTSG101#2, siVPS28#1 or siV2PS8#2), as compared to control cells (treated with non-targeting siRNAs, Ctrl#1 or #2) in HEK293 cells. Vinculin used as a gel loading control. B Ingenuity Pathway Analysis (IPA) of microarray results, showing top canonical pathways identified by annotation of genes whose expression was significantly (FDR < 0.05) downregulated or upregulated in cells depleted of ESCRT-I using two single siRNAs for each component, as compared to control cells. Blue asterisks indicate annotations related to metabolism of amino acids and fatty acids. Microarray data analysis was performed based on three independent experiments. C Gene ontology (GO) analysis of top biological processes identified by annotation of genes detected in microarray experiments as those with strongly downregulated expression (log2 fold change ≤ − 0.6; FDR < 0.05) upon ESCRT-I removal. D Heatmap visualizing expression of genes annotated to “small molecule metabolic process” (GO:0044281), whose mRNA levels were detected by microarray as downregulated after ESCRT-I removal. The genes encoding enzymes involved in oxidation of amino acids or fatty acids indicated with orange and green rectangles. E qPCR results showing the expression of genes encoding the indicated oxidative metabolism enzymes in cells lacking ESCRT-I, as compared to control cells, presented as fold changes with respect to averaged values measured for siCtrl#1 and #2 cells (siCtrl average). Mean values (n = 5 ± SEM) are presented. F Representative western blots (left panel), performed on the same samples as blots presented in A, showing the levels of the indicated oxidative enzymes in control or ESCRT-I-deficient cells. The graph (right panel) shows protein levels as fold change with respect to averaged values measured for control cells (siCtrl average) by densitometry analysis of western blotting bands. Vinculin was used as a gel loading control. Values derived from independent experiments and their means (n = 4 ± SEM) are presented. All the analyses shown in A–F were performed at three days post transfection with siRNAs (3 dpt). Statistical significance tested by comparison to siCtrl average. #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Among genes whose expression was commonly altered in samples with TSG101 or VPS28 depletion, we detected 855 genes with significantly upregulated expression and 820 with significantly downregulated expression, as compared to control cells (Fig. 1B). These genes were functionally annotated using the Ingenuity Pathway Analysis. As expected from our previous studies [14, 29, 30], ESCRT-I depletion led to elevated expression of many genes involved in inflammation and stress response (Fig. 1B).
Importantly, we observed that among genes with reduced expression in cells lacking ESCRT-I were those annotated to oxidative breakdown of amino acids and fatty acids (Fig. 1B). Our further analysis using Gene Ontology Biological Processes database, focusing only on 328 genes with the strongest downregulation (log2 fold change ≤ − 0, 6), showed that ESCRT-I depletion led to reduced expression of over 50 genes involved in “small molecule metabolism” (Fig. 1C–D). This list included mainly genes encoding enzymes responsible for oxidative metabolism of carboxylic acid-containing molecules such as fatty acids and amino acids (Fig. 1D, Table S1). Given that these genes encode enzymes involved in oxidation of such molecules, we refer to them hereafter as “amino acid or fatty acid oxidation genes”.
To validate the transcriptomic results, we focused on genes encoding IDH1 and DECR1 enzymes, that participate in fatty acid metabolism [41, 42], as well as SUOX, ALDH6A1 and BCKDHB [43,44,45] that are involved in amino acid metabolism. By quantitative RT-PCR (qRT-PCR), we independently confirmed the reduced mRNA levels of each of these enzymes in HEK293 cells lacking TSG101 or VPS28 (Fig. 1E). However, siTSG101#1, that was less efficient in depleting TSG101 and VPS28 proteins than siTSG101#2 (shown in Fig. 1A), had a weaker effect on the mRNA levels of the analyzed enzymes. This suggested that high efficiency of TSG101 depletion has to be achieved to lower the expression of amino acid or fatty acid oxidation genes. To investigate whether reduced expression of the chosen genes occurred also in other cell types, we performed qRT-PCR analysis in HepG2 hepatoblastoma cell line with siRNA-mediated removal of TSG101 or VPS28 proteins using siTSG101#2 and siVPS28#1 (Fig. S1A) or CRISPR-Cas9-mediated knock-out of TSG101 gene (Fig. S1B–C). The efficiencies of using siRNAs or CRISPR-Cas9 system in these cells were assessed by qRT-PCR (Fig. S1A) or western blotting (Fig. S1B), respectively. In each case, deficiency of ESCRT-I complex led to reduced expression of analyzed amino acid or fatty acid oxidation genes (Fig. S1A and C).
To address whether changes in the expression of amino acid or fatty acid oxidation genes observed upon ESCRT-I deficiency could affect cell metabolism, we tested protein levels of chosen enzymes in HEK293 cells by western blotting. We confirmed that siRNA-mediated depletion of TSG101, using siTSG101#1, or VPS28, using siVPS28#1 or #2, at 3 dpt caused reduced abundance of IDH1, SUOX and ALDH6A1 oxidative enzymes (Fig. 1F). Thus, the transcriptomic analysis and its validation demonstrated that ESCRT-I deficiency causes reduced gene expression and protein levels of enzymes involved in oxidation of small molecules such as amino acids and fatty acids.
ESCRT-I deficiency causes intracellular accumulation of lipids, including phospholipids that accumulate in the enlarged ERReduced expression of genes encoding enzymes of fatty acid oxidation is often associated with intracellular accumulation of lipids [46]. Moreover, we observed increased expression of several genes encoding enzymes of biosynthesis of various types of lipids (fatty acids, triglycerides or phospholipids) in cells lacking ESCRT-I (Fig. S2A). Thus, we investigated the abundance of such lipids in cells with TSG101 or VPS28 depletion using single siRNAs (siTSG101#2 and siVPS28#2) against each protein (Fig. 2A).
Fig. 2
Cells lacking ESCRT-I have elevated levels of lipids and enlarged ER. A Western blots showing the efficiencies of siRNA-mediated depletions of ESCRT-I subunits, TSG101 or VPS28 (cells treated with siTSG101#2 or siV2PS8#2), as compared to control conditions (two non-targeting siRNAs, Ctrl#2 or #3) in HEK293 cells. Vinculin used as a gel loading control. B Results of gas chromatography followed by mass spectrometry (GC–MS) showing intracellular levels of free fatty acids—FFA, triglycerides—TG, diacylglycerides—DAG or phospholipids—PL (shown as nmol/µg of proteins) in control or ESCRT-I-depleted cells. Values derived from independent experiments and their means (n = 4 ± SEM) are presented. Values for siCtrl average are averaged values measured for cells transfected with siCtrl#2 or siCtrl#3. C–D Maximum intensity projection confocal images of live control or ESCRT-I-depleted cells. The images show the intracellular distribution of neutral lipids—NL (green), or phospholipids—PL (red) stained with Nile Red dye (shown in C) as well as the intracellular distribution of NLs stained with BODIPY 493/503 and the ER stained with ER-tracker (green or red, respectively in D). The dot plots show total fluorescence intensities per cell (expressed in arbitrary units, a.u.), as compared to averaged values measured for cells transfected with siCtrl#2 or siCtrl#3 (siCtrl average). Average number of cells analyzed per condition was 2936 for siCtrl#2, 2768 for siCtrl#3, 1419 for siTSG101#2 and 1871 for siVPS28#2. E Maximum intensity projection confocal images of fixed control or ESCRT-I-depleted cells stained using antibodies recognizing CLNX protein (red) or mono- and polyubiquitinated protein conjugates (Ub; green). The dot plots show total fluorescence intensities per cell (expressed in arbitrary units, a.u.), as compared to averaged values measured for cells transfected with siCtrl#2 or siCtrl#3 (siCtrl average). Average number of cells analyzed per condition was 4414 for siCtrl#2, 4481 for siCtrl#3, 2126 for siTSG101#2 and 2473 for siVPS28#2. Cell nuclei in C–E marked with Hoechst stain (blue). Scale bars, 50 μm. Dot plot values in C–E derived from independent experiments (dots) and their means (n = 3 ± SEM) are presented. All the analyses shown in B–E were performed at three days post transfection with siRNAs (3 dpt). Statistical significance tested by comparison to the siCtrl average values. #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001
To assess the intracellular content of fatty acids, we performed gas chromatography followed by mass spectrometry (GC–MS) (Fig. 2B). We measured free fatty acids (FFAs) and fatty acid-containing lipids, i.e., triglycerides (TGs), diacylglycerides (DAGs) and phospholipids (PLs). Depletion of TSG101 or VPS28 led to slightly elevated levels of FFAs and TGs and to an even stronger increase in membrane lipids, DAGs and PLs (Fig. 2B). To verify elevated lipid levels, we stained control and ESCRT-I-depleted cells with Nile Red dye (NR) and imaged them by confocal microscopy. NR emits green fluorescence, when bound to neutral lipids (NLs, that include cholesterol or TGs), or red fluorescence, when bound to PLs [47]. Consistent with the GC–MS results, we observed that lack of ESCRT-I led to increased levels of both NLs and PLs (Fig. 2C). We confirmed elevated abundance of NLs in cells lacking TSG101 or VPS28 by using BODIPY 493/503 dye (Fig. 2D) that emits green fluorescence when bound to these lipids [48]. The amounts of NLs were increased primarily in lipid droplets—LDs (Fig. 2C–D), identified as punctate structures positive for both NLs and PLs [49]. The LDs were more abundant upon ESCRT-I depletion (Fig. S2B), but had similar mean size as in control cells (Fig. S2C). PLs accumulated in ESCRT-I-deficient cells also outside of LDs, in a region resembling the ER by shape and localization (Fig. 2C). As PLs are the most abundant lipids in the cell, that build cellular membranes, we reasoned that their accumulation in the ER region could reflect an increase of ER volume. Indeed, we noticed that cells lacking TSG101 or VPS28 had a strongly increased ER content as measured by confocal microscopy using ER-tracker Red dye (Fig. 2D) and antibodies recognizing calnexin (CLNX), an ER-resident protein (Fig. 2E).
In mammalian cells the ER size is restricted by its selective autophagic degradation that occurs in an ubiquitin-dependent manner [50]. To address whether the enlargement of the ER due to lack of ESCRT-I could be a consequence of an impaired ER degradation, we quantified the amount of ubiquitin on the CLNX-positive compartment. Similarly as we reported for RKO colon cancer cells [14], depletion of TSG101 or VPS28 in HEK293 cells led to a strong intracellular accumulation of ubiquitin (Fig. 2E), likely due to the impairment of multiple ESCRT-I-mediated degradation processes [14]. Some of the accumulated ubiquitin was enriched in the CLNX-positive compartment (Fig. 2E) indicating that ESCRT-I deficiency may impair the ER-phagy.
Hence, we discovered that lack of TSG101 or VPS28 leads to increased abundance of FFAs and lipids, such as PLs that accumulate in intracellular membranes including the enlarged ER. Although increased amount of fatty acid-containing lipids in cells lacking ESCRT-I could be in part due to their impaired lysosomal degradation, for instance ER-phagy inhibition, it could also result from reduced oxidative breakdown of FFAs and increased lipid biosynthesis, as implied by the changes in gene expression described above (Fig. 1 and S2).
ESCRT-I deficiency does not impair mitochondrial biogenesis or ATP-linked mitochondrial respirationBy GO Cellular Component analysis of genes with strongly reduced expression in cells lacking TSG101 or VPS28, we identified many genes that encode mitochondrial proteins (Fig. S3A, Table S2; around 50 genes that included many of the amino acid or fatty acid oxidation genes indicated in Fig. 1D). Reduced expression of genes encoding mitochondrial proteins could potentially affect mitochondrial biogenesis. However, we did not observe a general reduction in the expression of genes encoding structural mitochondrial proteins or core components of the citric acid cycle or oxidative phosphorylation. To investigate the consequence of ESCRT-I deficiency on the abundance of mitochondria, we performed confocal microscopy analysis of HEK293 cells stained with MitoTracker Deep Red FM dye. We observed that MitoTracker staining was not reduced in the absence of ESCRT-I at 3 dpt, on the contrary, it was increased (Fig. 3A). These data indicated that ESCRT-I deficiency did not impair the biogenesis of mitochondria.
Fig. 3
The abundance of functional mitochondria and the rate of ATP-linked mitochondrial respiration are not reduced upon ESCRT-I deficiency. A–B Maximum intensity projection confocal images of live control (treated with non-targeting siRNAs, Ctrl#2 or #3) or ESCRT-I-depleted HEK293 cells (treated with siTSG101#2 or siV2PS8#2). The images show the intracellular content and distribution of all mitochondria stained with MitoTracker Deep Red FM dye (red in A) and of mitochondria with proper membrane potential stained with Tetramethylrhodamine, Ethyl Ester (TMRE, red in B). Cell nuclei marked with Hoechst 33342 stain (blue). Scale bar, 50 μm. The dot plots show total fluorescence intensities per cell (expressed in arbitrary units, a.u.), as compared to averaged values measured for cells transfected with siCtrl#2 or siCtrl#3 (siCtrl average). Values derived from independent experiments (dots) and their means (n = 3 ± SEM) are presented. Average number of cells analyzed per condition was 2542 for siCtrl#2, 2527 for siCtrl#3, 1464 for siTSG101#2 and 1729 for siVPS28#2. C A representative time-series graph (left) showing changes of oxygen consumption rate (OCR) with time (pmol/min) in control or ESCRT-I-depleted cells upon: basal respiration (untreated cells; time-points 1–3), inhibition of ATP-linked respiration (oligomycin treatment; time-points 4–6), maximal respiration (FCCP treatment; time-points 7–9) and non-mitochondrial respiration (antimycin A and rotenone treatment; time-points 10–12). The bar graph (right) shows the intensity of the indicated processes calculated based on the results shown in the time-series graph. The results shown in both graphs were normalized to cell number reflected by DNA staining with Hoechst 33342 dye. Mean values in both graphs derived from technical repetitions of one experiment (n = 4 or 5 ± SEM) are presented. D Maximum intensity projection confocal images of fixed control, ESCRT-I-depleted, OPA1-depleted or DRP1-depleted cells stained using antibodies recognizing TOM20 protein (red). Cell nuclei marked with DAPI stain (blue). Scale bar, 50 μm. The dot plot (bottom left) shows total fluorescence intensity per cell (expressed in arbitrary units, a.u.), as compared to averaged values measured for cells transfected with siCtrl#2 or siCtrl#3 (siCtrl average). Values derived from independent experiments (dots) and their means (n = 3 ± SEM) are presented. Graph on the right shows percentage of cells with fragmented, regular or tubular mitochondria. Average number of cells analyzed per condition was 1304 for siCtrl#2, 1132 for siCtrl#3, 598 for siTSG101#2, 765 for siVPS28#2, 1170 for siDRP1 and 896 for siOPA1. All the analyses shown in A-D were performed at three days post transfection with siRNAs (3 dpt). Statistical significance tested by comparison to the siCtrl average values. #P < 0.1, *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001
As ESCRTs are required for mitochondria degradation in lysosomes [26, 27], the accumulation of mitochondria observed in cells lacking TSG101 or VPS28 could be a result of impaired autophagic removal of damaged mitochondria. Hence, to address whether ESCRT-I deficiency affects the functionality of mitochondria, we measured mitochondrial membrane potential using Tetramethylrhodamine Ethyl Ester (TMRE) dye that stains properly functioning mitochondria. By confocal microscopy, we observed that the TMRE fluorescence intensity was slightly reduced upon TSG101 depletion and not altered upon VPS28 depletion (Fig. 3B). Hence, the overall mitochondrial membrane potential did not increase upon ESCRT-I deficiency (Fig. 3B) as it was the case for overall mitochondria content (shown in Fig. 3A). This analysis suggested that cells lacking ESCRT-I retain functional mitochondria but also accumulate excess mitochondria that do not have proper membrane potential.
To address whether the reduced expression of amino acid or fatty acid oxidation genes or the accumulation of damaged mitochondria upon ESCRT-I deficiency affect mitochondrial respiration, we measured oxygen consumption rate (OCR) using the Agilent Seahorse XFe24 Analyzer. Control HEK293 cells showed typical OCR curve, indicating basal respiration (as a sum of ATP-linked and proton leak-related processes) that was lower than maximal respiratory capacity (Fig. 3C). However, cells lacking ESCRT-I components had elevated basal respiration, reaching maximal capacity, due to increased proton leak (Fig. 3C). The elevated proton leak was consistent with the above-described accumulation of damaged mitochondria upon ESCRT-I deficiency (shown in Fig. 3A–B). Importantly, the OCR linked to ATP production was not impaired in cells lacking ESCRT-I depletion (Fig. 3C). It was not affected by depletion of TSG101 and was even elevated upon VPS28 depletion as compared to control cells (Fig. 3C).
Overall, ESCRT-I deficiency does not affect the abundance of mitochondria with proper membrane potential. Moreover, despite strongly impaired lysosomal degradation of proteins [14, 30] and lipids (shown in Fig. 2B–C), as well as reduced levels of enzymes involved in amino acid and fatty acid degradation (shown in Fig. 1F), ESCRT-I depletion does not impair ATP-dependent mitochondrial respiration. Hence, the observed metabolic changes in cells lacking ESCRT-I are not due to general mitochondria malfunction.
Lack of ESCRT-I promotes fragmentation of mitochondriaAnalyzing the confocal microscopy images after MitoTracker staining (shown in Fig. 3A), we noticed that cells lacking TSG101 or VPS28 may have altered morphology of mitochondria. To address this in detail, we analyzed by confocal microscopy the intracellular distribution of a mitochondrial marker, TOM20 protein in HEK293 cells. Consistent with higher mitochondrial abundance in ESCRT-I-deficient cells, that we observed using MitoTracker, these cells had elevated staining intensity of TOM20 as compared to control cells (Fig. 3D). Of note, the quantitative analysis of the confocal images pointed out that in part accumulation of ubiquitin in cells lacking ESCRT-I occurs on the TOM20-positive compartment (Fig. S3B). However, as we did not observe clear colocalization of TOM20 with ubiquitin accumulated in cells lacking ESCRT-I (Fig. S3B), we could not conclude that the elevated signal intensity of MitoTracker (shown in Fig. 3A) or TOM20 (Fig. 3D) in cells lacking ESCRT-I is due to accumulation of non-degraded, ubiquitinated mitochondria.
Mitochondrial morphology is determined by fusion and fission processes mediated by a number of regulators including dynamin-related protein 1 (DRP1), the master regulator of mitochondrial fission, and optic atrophy 1 (OPA1) protein, that facilitates mitochondrial fusion [51]. In order to assess how lack of ESCRT-I affects the morphology of mitochondria, we used the supervised machine learning module of the software dedicated for the Opera high-content screening microscope, with which we obtained the confocal images. This allowed us to compare the distribution of TOM20 protein in cells lacking TSG101 or VPS28 to TOM20 distribution in cells lacking DRP1 or OPA1, based on a large number of confocal images. According to this analysis, most of control HEK293 cells had regularly shaped mitochondria with only around 10% of cells with fragmented mitochondria identified (Fig. 3D). As expected, siRNA-mediated depletion of OPA1 caused mitochondrial fragmentation (over 90% of cells), whereas most of the cells with depletion of DRP1 had tubular mitochondria and barely any cells lacking DRP1 (around 1%) had fragmented mitochondria (Fig. 3D). Interestingly, ESCRT-I deficiency caused an increased percentage of cells (50%-70%) identified as those containing fragmented mitochondria (Fig. 3D).
These data indicated that the presence of functional ESCRT-I promotes mitochondria fusion or inhibits mitochondria fission. The molecular mechanisms underlying this regulation are yet to be discovered.
Cells lacking ESCRT-I activate glycolytic metabolismSmall mitochondria are characteristic for cells that base their metabolism on aerobic glycolysis [52, 53]. Hence, we reasoned that the reduced expression of amino acid or fatty acid oxidation genes in ESCRT-I-deficient cells could be associated with changes in expression of genes involved in glucose catabolism. Although the Ingenuity Pathway Analysis of genes with commonly induced expression upon TSG101 or VPS28 depletion (Fig. 1B) did not indicate such annotation, we investigated in more detail the transcriptomic results focusing on genes encoding enzymes involved in glycolysis, hence metabolism of glucose to pyruvate (Fig. 4A), and in conversion of pyruvate to acetyl-CoA (Fig. S4A). We found that ESCRT-I deficiency caused elevated expression of several genes that encode glycolytic enzymes (Fig. 4A) but had no particular effect on the expression of genes responsible for pyruvate to acetyl-CoA conversion (Fig. S4A). By qRT-PCR analysis performed at 3 dpt (the same time-point as of the transcriptomic analysis), we verified the increased expression of HK2, ENO2, PFKFB3 and PFKP glycolytic enzymes in HEK293 cells upon depletion of VPS28 with two independent siRNAs (Fig. S4B). However, such increase was not observed in cells transfected with siTSG101#1 and occurred only for ENO2 and PFKFB3 in cells transfected with siTSG101#2 (Fig. S4B).
Fig. 4
ESCRT-I dysfunction leads to increased aerobic glycolysis and other metabolic changes reminiscent of the Warburg effect. A Heatmap visualizing microarray results regarding the expression of genes encoding enzymes involved in glycolysis in HEK293 cells after removal of ESCRT-I using two siRNAs for each component (siTSG101#1 or siTSG101#2, siVPS28#1 or siVPS28#2), as compared to control cells (treated with non-targeting siRNAs, Ctrl#1 or #2). Microarray data analysis was performed based on three independent experiments at three days post transfection with siRNAs (3 dpt). B qPCR results showing the expression of genes encoding glycolytic enzymes in cells depleted of TSG101 or VPS28 using single siRNAs (siTSG101#2 or VPS28#2), as compared to control cells (treated with non-targeting siRNA, Ctrl#2) at 4 dpt. The mean values (n = 4 ± SEM) presented as fold changes with respect to values for control cells. C Representative western blots (left panel) showing the levels of TSG101, VPS28 and the indicated glycolytic enzymes in control (treated with non-targeting siRNAs, Ctrl#2 or #3) or ESCRT-I-depleted (siTSG101#2 or siVPS28#2) cells. The graph (right panel) shows protein levels as fold change with respect to averaged values measured for siCtrl#2 and #3 assessed by densitometry analysis of western blotting bands. Vinculin was used as a gel loading control. Mean values (n = 4 ± SEM) are presented. D Intracellular levels of lactate (shown as nmol/µg of DNA) in control (siCtrl#1 or #2) or ESCRT-I-deficient cells. Mean values (n = 3 ± SEM) are presented. Values for siCtrl average are averaged values measured for cells transfected with siCtrl#1 or siCtrl#2. E A representative time-series graph (left) showing changes of extracellular acidification rate (ECAR) with time (mpH/min) in control (siCtrl#2 or #3) or ESCRT-I-depleted cells at 4 dpt upon: basal respiration (untreated cells; time-points 1–3), inhibition of ATP-linked respiration (oligomycin treatment; time-points 4–6), maximal respiration (FCCP treatment; time-points 7–9), non-mitochondrial respiration (antimycin A and rotenone treatment; time-points 10–12) and inhibition of glycolysis (2-DG treatment; time-points 13–15). The bar graph (right) shows the intensity of the indicated processes calculated based on the results shown in the time-series graph. The results shown in both graphs were normalized to cell number reflected by DNA staining with Hoechst 33342 dye. Mean values in both graphs derived from technical repetitions of one experiment (n = 5 ± SEM) are presented. F–G Abundance of chosen metabolites detected by 1H-NMR analysis in the medium from control or ESCRT-I-deficient cells as compared to levels in fresh DMEM (n = 4 ± SEM). The medium was collected after 24 h of cell culture from 3 to 4 dpt. H Intracellular levels of chosen amino acids detected by 1H-NMR analysis of metabolites in pellets of control or ESCRT-I-deficient cells at 4 dpt (n = 4 ± SEM). Statistical significance in B, C, F, G and H tested by comparison to siCtrl#2, whereas in D and E tested by comparison to siCtrl average. #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Reasoning that 3 dpt could be too early to observe a full effect on glycolytic gene expression in cells lacking TSG101, we performed the analysis in cells transfected with siTSG101#2 or siVPS28#2 at 4 dpt and observed a prominent increase in the expression of all analyzed glycolytic genes (Fig. 4B). As verified by western blotting, depletion of TSG101 or VPS28 at 4 dpt was very efficient (Fig. 4C). The upregulated expression of genes encoding ENO2, PFKFB3 and PFKP (but not HK2) also occurred in ESCRT-I-deficient HepG2 cells at 3 dpt (Fig. S4C–D). As in HEK293 cells, we observed a stronger increase in the expression of these glycolytic genes in HepG2 cells with siRNA-mediated VPS28 depletion than upon TSG101 depletion (Fig. S4C). However, CRISPR-Cas9-mediated TSG101 depletion led to clearly upregulated expression of genes encoding ENO2, PFKFB3 and PFKP (Fig. S4C).
Next, we addressed whether increased expression of genes encoding particular glycolytic enzymes translates into their higher abundance and increased glycolytic metabolism. Analyzing the cells at 4 dpt, we observed elevated protein levels of HK2 and PFKFB3 enzymes upon depletion of both VPS28 or TSG101 (Fig. 4C). To address whether ESCRT-I-deficient HEK293 cells activate glycolytic metabolism, we measured the intracellular content of lactate, the product of anaerobic glucose metabolism. We found that at 4 dpt, cells lacking TSG101 or VPS28 had clearly elevated intracellular levels of lactate (Fig. 4D). To verify the effect of ESCRT-I deficiency on aerobic glycolysis, we used the Agilent Seahorse XFe24 Analyzer to measure the extracellular acidification rate (ECAR) that is increased upon release of lactate [54]. This analysis showed that, as compared to control cells, cells lacking TSG101 or VPS28 had elevated ECAR, primarily due to glycolysis (Fig. 4E), although their non-glycolytic acidification was also slightly elevated (Fig. 4E). Of note, depletion of ESCRT-I components for 4 days caused an increase of both, basal glycolysis as well as maximal glycolytic capacity (Fig. 4E).
Hence, in cells lacking ESCRT-I, the inhibited expression of amino acid or fatty acid oxidation genes is associated with activated expression of glycolytic genes and induction of glycolytic metabolism. These results suggest that functional ESCRT-I may promote oxidative metabolism of lysosome-derived nutrients over glycolytic metabolism.
Metabolic reprogramming upon ESCRT-I deficiency resembles the Warburg effectCollectively, the above-described results suggested that lack of ESCRT-I may lead to metabolic reprogramming similar to the Warburg effect that is characterized by increased consumption of glucose and some amino acids [10]. To verify this, we investigated the effect of ESCRT-I deficiency on consumption of metabolites using nuclear magnetic resonance (1H-NMR) approach. We analyzed the changes in abundance of metabolites in the medium of cells during 24 h of culture, from 3 to 4 dpt, as well as assessed the intracellular metabolite levels at 4 dpt (Tables S3–S6). 1H-NMR allowed to detect various extracellular and intracellular metabolites including most of the amino acids (Fig. S5A-B, Tables S3 and S4). However, glucose and pyruvate were detected only in the medium but not inside the cells (Fig. S5A–B, Tables S3 and S4). We observed that control cells consumed some glucose from the medium but also released pyruvate and lactate (Fig. 4F). Importantly, cells lacking TSG101 or VPS28 had increased glucose consumption, lower pyruvate release and higher lactate release (Fig. 4F), indicating elevated conversion of medium-derived glucose through pyruvate into lactate.
Upon the Warburg effect, elevated glucose consumption in cells is associated with increased uptake of glutamine, serine and glycine. We observed that depletion of TSG101 or VPS28 led to increased usage of glutamine and serine but not glycine from the medium (Fig. 4G). Moreover, cells lacking ESCRT-I had reduced intracellular abundance of glutamine, glycine and (less significantly) serine (Fig. 4H). Lower medium and/or intracellular levels of these amino acids suggested their increased utilization. Of note, ESCRT-I deficiency had no effect on extracellular or intracellular levels of BCAAs (leucine, isoleucine and valine) (Fig. S5C–D), showing that elevated glutamine, serine and glycine consumption was not due to general alterations in amino acid metabolism.
The increased glutamine consumption occurs in cells with the Warburg effect, among other purposes, to maintain ATP production, that would otherwise be impaired due to shift to aerobic glycolysis [5]. Accordingly, our 1H-NMR analysis showed that lack of TSG101 or VPS28 did not affect the intracellular ATP or ADP levels, as compared to control cells (Fig. S5E). This was consistent with the OCR analysis which showed that ESCRT-I deficiency did not impair ATP-linked oxidation (shown in Fig. 3C). Among molecules biosynthesized from one-carbon metabolism of glycine and serine, cells exerting the Warburg effect produce high amounts of glutathione [55]. Consistently, we observed elevated glutathione abundance in cells lacking TSG101 or VPS28 (Fig. S5F).
Hence, we confirmed that ESCRT-I dysfunction leads to metabolic reprogramming resembling the Warburg effect. Noteworthy, this reprogramming is more pronounced in cells lacking VPS28 than in cells lacking TSG101 (Fig. 4F–H), potentially due to faster response in upregulation of glycolytic gene expression upon VPS28 depletion (shown in Fig. S4B and 4B).
mTORC1 signaling is not implicated in regulation of cell metabolism upon ESCRT-I depletionNext, we sought to investigate which signaling pathways could be involved in the altered expression of metabolic genes upon ESCRT-I depletion. Cell metabolism is largely controlled by mTORC1 signaling that regulates amino acid, fatty acid and glucose metabolism. We have previously found that in RKO colorectal cancer cells, ESCRT-I deficiency does not affect general mTORC1 signaling, for instance phosphorylation of mTOR kin
留言 (0)