
記住我

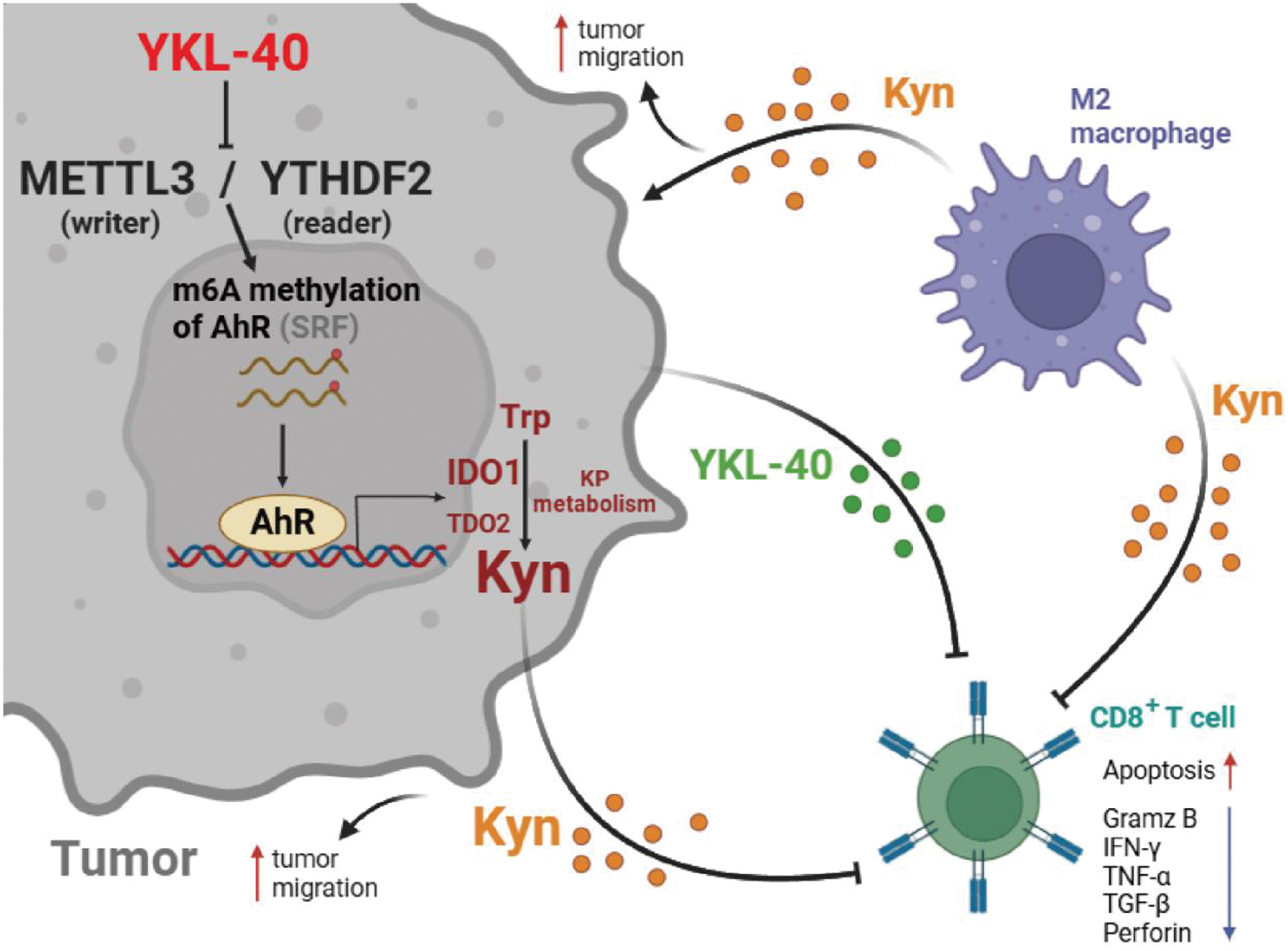






We generated multiple clones from a single donor using Sendai virus to investigate the heterogeneity among hiPSC clones (Figs. 1A and S1A). Approximately 20 colonies were observed, and individual colonies were manually picked and subsequently expanded on Matrigel-coated culture plates. The resulting six clones (CL1-6: clone 1–6) formed stable colonies devoid of differentiated cells and displayed typical hiPSC morphology [7], including prominent nucleoli and distinct borders (Fig. 1B). All clones were maintained under the same conditions to minimize the effect of subculture processes. Immunofluorescence confirmed the expression of pluripotency-associated markers, including SOX2, SSEA4, and OCT4 in all clones (Figs. 1C and S1B). The protein levels of SOX2 and OCT4 were not significantly different among the clones (Fig. 1D, F, and G). In addition, the six clones exhibited similar proliferation rates (~ 60%) and normal karyotypes (Figs. 1E, H, and S1C). In summary, we obtained six hiPSC clones that expressed pluripotency markers and exhibited comparable growth rates.
Fig. 1
Generation and characterization of human induced pluripotent stem cell (hiPSC) clones. A Experimental design for the evaluation of clonal heterogeneity in hiPSC. B Phase-contrast images of the clones. Scale bar = 50 μm. C Representative immunostaining images for SOX2 (green), SSEA4 (red), OCT4 (yellow), and Hoechst (blue) staining of the clones. Scale bar = 50 μm. D Representative immunoblotting images for OCT4, SOX2, and H3. E Representative images for 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay. Scale bar = 50 μm. F–G Quantification of signal intensity of the immunoblotting for OCT4 (F) and SOX2 (G). Data show two technical replicates per group, and all values were normalized to H3. H Quantification of EdU + proliferating cells stained with Hoechst. Data show six technical replicates per group. All statistical analysis were performed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (F, H) or Brown-Forsythe and Welch ANOVA with Dunnett’s multiple comparisons test (G). Data are presented as bar graphs and error bars with mean ± standard deviation
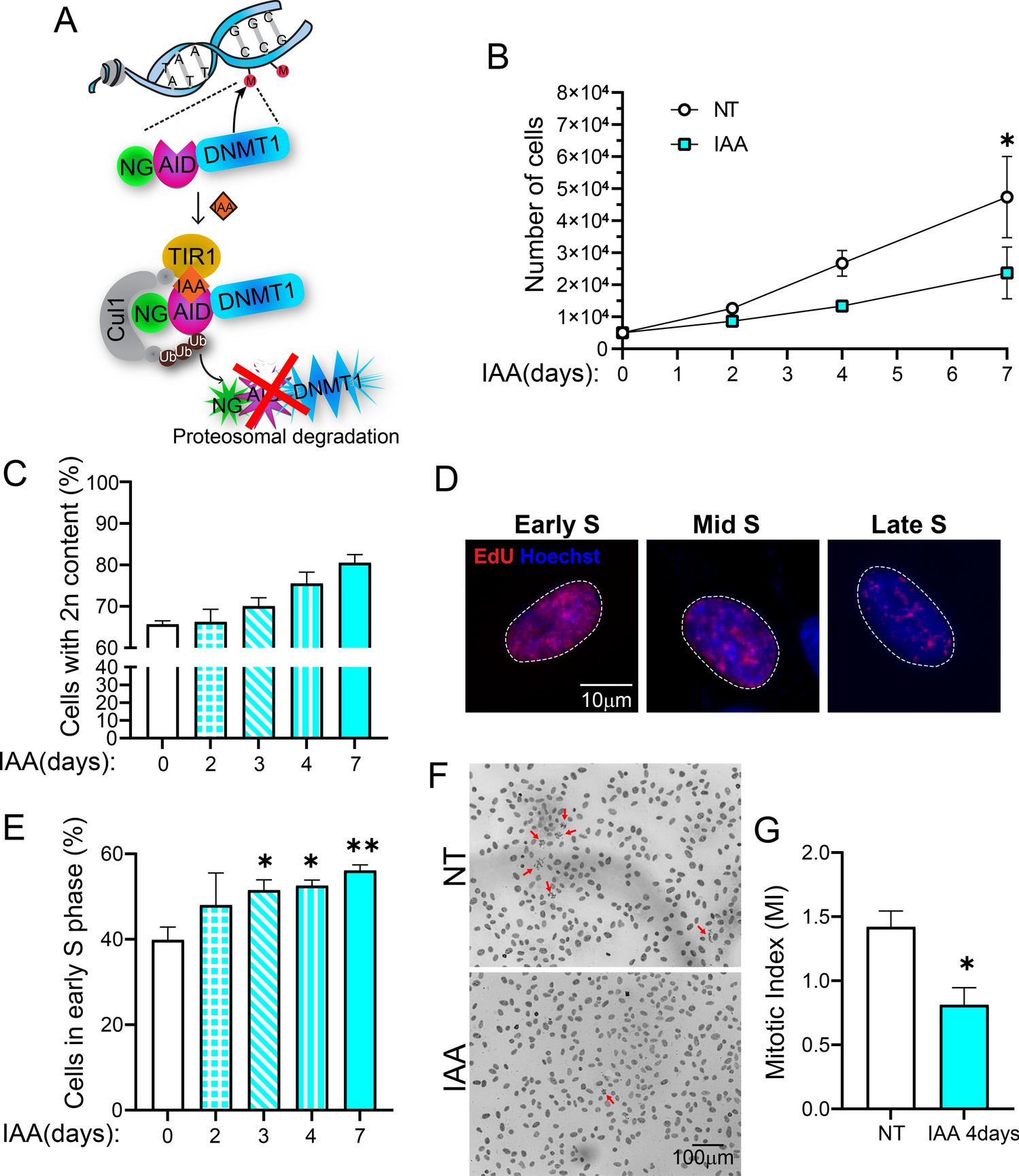
Differentiation capacity of cardiomyocytes varied among clonesTo assess the differentiation potential of the clones, we induced cardiomyocytes (CMs) from six hiPSC clones following an established protocol [14]. All clones were simultaneously treated with the same concentration of CHIR99021 (5 µM), a GSK-3 inhibitor. The proportion of TNNT2 (cardiac troponin-T)-positive cells were compared via flow cytometry between day 12 to 15 of differentiation (Fig. 2A). CL1 and 3 consistently exhibited robust differentiation rates (> 60%), whereas CL2 showed a lower differentiation rate (< 50%). CL4, 5, and 6 showed variability among batches, thereby these clones were excluded in further analysis (Fig. 2B, C, and S2A). The differentiated cells derived from CL1 and 3 showed spontaneous beating by day 15 (white arrows in Fig. 2D), whereas CL2 showed a higher proportion of non-beating cells (Videos S1 and 2). Immunofluorescence confirmed the coexistence of CMs (TNNT2-positive) and non-CMs (TNNT2-negative) in CL2 (Figs. 2E and S2B). Using quantitative reverse transcription-polymerase chain reaction (qRT-PCR), we compared the expression levels of CM markers (TNNT2 and MYH7/6: myosin heavy chain 7/6) and a non-CM marker (VIM: vimentin) (Fig. 2F). VIM was upregulated in CL2, indicating the presence of a mixed proportion of CMs and non-CMs. To mitigate the potential effects of CHIR99021 concentration, differentiation was also induced at 4 and 6 μM. At 4 μM of CHIR99021, the results were consistent with previous observations, while differentiation did not occur in any of the clones at 6 μM of CHIR99021 (Fig. S3A–C and Videos S3–S5).
Fig. 2
Evaluation of the differentiation efficiency and functionality of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). A Experimental scheme for the evaluation of cardiac differentiation capacity. B Flow cytometry of cardiac troponin T (TNNT2) expression in hiPSC-CMs showed that the clones had different efficiencies, based on which they were divided into two groups—PC (blue) and NPC (red). Three or four technical replicates per group are shown. C Representative flow cytometric histograms showing the TNNT2 + (%) cells of hiPSC-CMs when hiPSC clones (CL1- 3) were treated with 5 µM of CHIR99021. Gray peaks represent the non-stained control. D Phase-contrast images of hiPSC-CMs derived from PC (left, CL1) and NPC (right, CL2); beating CMs were represented with white arrows. Scale bar = 1000 μm. E Representative images of Immunostaining for TNNT2 (green) and Hoechst (blue) staining of hiPSC-CMs (day 15). F Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) of CM (TNNT2, MYH7, and MYH6) and non-CM (VIM) markers in hiPSC-CMs. All values were normalized to GAPDH. Data show three or four technical replicates per group. G Field potential analysis of hiPSC-CMs using a multielectrode (MEA) system. Quantification of the beating rate, spike amplitude, and spike slope. Data represent three technical replicates per group. H Representative heatmap images of conduction map. The cardiac beating begins where the plot is blue (shortest delay from the beat origin) and ends where the plot is red (longest delay from the beat origin). All statistical analysis were performed using unpaired Student’s t-tests or Welch’s t-tests; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are presented as bar graphs and error bars with mean ± standard deviation (B, G) or standard error of mean (F)
Functional assessment of CMs was performed using a multielectrode array system, and data were acquired using CMs at day 20, which was 5 days post-plating. We detected active beating of CMs in CL1 and 3, but not in CL2 (Fig. S3D), possibly due to the low proportion of CMs (Fig. 2B-F). There was no significant difference of beating rate between CL1 and 3, whereas CL3 exhibited an increase in spike amplitude and slope (Fig. 2G and S3D). The conduction map showed a delay in conduction, which was no significant difference of conduction velocity, maximum delay, and propagation consistency between CL1 and 3, with no observed arrhythmias (Fig. 2H and S3E). Recent studies have reported that hiPSC-derived CMs show conduction velocities ranging from 0.04 m/s to 1.0 m/s [15, 16], and CL1 and 3 showed the velocities within this range except for CL2. Overall, the hiPSC clones (CL1-3) were classified into two groups based on their differentiation capacity, with CL2 showing a low capacity, resulting in mixed proportions of CMs and non-CMs—CL1 and 3 as productive clone (PC) and CL2 as non-productive clone (NPC).
Phosphorylation state of ERK1/2 signaling pathway was elevated in NPCSignaling pathways such as Wnt, BMP, and ERK play crucial roles in cardiac development [17, 18]. Recent studies have highlighted the effect of the phosphorylation status of signaling kinases in hiPSCs on their differentiation potential [19, 20]. To compare the signaling profiles of the hiPSC clones (PC and NPC), we conducted a human phospho-kinase array. We examined whether the phosphorylation levels of Wnt/β-catenin or GSK-3α/β differed between the two groups, because we used a GSK-3 inhibitor, a regulator of the Wnt/β -catenin pathway, to induce CM differentiation (Fig. 2A). However, the phosphorylation of GSK-3α/β (Ser21/Ser9) did not show significant differences among the clones (Fig. 3A and B). We also assessed the nuclear translocation of β-catenin using immunofluorescence, which revealed no discernible differences across all clones (Fig. S4A).
Fig. 3
The phosphorylation state of ERK1/2 signaling pathway was elevated in non-productive clone (NPC). A Chemiluminescent images of human phosphokinase array revealed distinct phosphorylation levels of ERK1/2 (T202/Y204 and T185/Y187), eNOS (S1177), p38α (T180/Y182), and p53 (S46) among clones (CL1-3). B Mean pixel density of 37 kinase phosphorylation sites and two related total proteins, normalized to reference spots. Statistical analysis was performed using multiple unpaired Student’s t-tests, and the two-stage step-up method of Benjamini, Krieger, and Yekutieli was used for the correction of multiple comparisons; *q < 0.05. Data represent two technical replicates per clone (PC: CL1 and 3, NPC: CL2). C Volcano plot of multiple unpaired Student’s t-tests showing that four phosphorylation sites were significantly different between PC and NPC; the largest difference is highlighted with a pink box. D Representative immunoblotting images for ERK1/2, phosphorylated ERK1/2 (T202/Y204), and H3. T: threonine, Y: tyrosine, S: serine
The phosphorylation levels of ERK1/2 (Thr202/Tyr204 and T185/Y187), p38α (Thr180/Tyr182), and eNOS (Ser1177) were significantly elevated in NPC, whereas those of p53 (Ser46) were increased in PC (Fig. 3B and C). Of these pathways, previous studies have reported that ERK1/2 [18] and p38α [21, 22] pathways are involved in CMs differentiation. The difference in the phosphorylation level of ERK1/2 between PC and NPC was the most pronounced in this study (Fig. 3C), and we confirmed the increased phosphorylation of ERK1/2 in NPC using immunoblotting (Fig. 3D). Taken together, the level of phosphorylated ERK1/2 was elevated in NPC compared to that in PC, whereas no differences were observed in the regulation of the Wnt pathway.
Distinct transcriptomic profiles revealed clonal heterogeneityRecent studies have reported that variations in the gene expression patterns of human pluripotent stem cells can affect their potential to differentiate into various cell lineages [23]. To analyze the transcriptomic profiles of the three clones (hiPSC #1 CL1-3), we performed bulk RNA-seq, followed by quality control of the raw read data (Fig. S5A). We compared our data with previously published data using principal component analysis (PCA) to ensure that all clones exhibited transcriptional signatures consistent with those of hiPSCs [24]. Our clones were closely clustered with hiPSCs, contrasting with other tissues, based on data from the Genotype-Tissue Expression (GTEx) portal (Fig. 4A).
Fig. 4
Transcriptomic analysis of human induced pluripotent stem cell (hiPSC) clones using bulk RNA-sequencing. A Principal component analysis (PCA) plot showing that generated hiPSC clones were in a cluster of previous iPSC data and distant from data from other tissues, based on data from the Genotype-Tissue Expression [GTEx] portal. B Heatmap displayed differentially expressed genes (DEGs, log2|FC|> 1 and adjusted P-values (Padj) < 0.05) between PC and NPC. C, D Gene Ontology (GO) analysis of DEGs in the biological process (BP), molecular function (MF), and cellular component (CC) categories. The emapplots (C) and bar plots (D) showed significantly enriched GOs in the two groups. The blue and red dots and bars indicate upregulated GOs in PC and NPC, respectively. E Chord diagram illustrating inter-relationships between the genes related to the GOs identified in C. F The volcano plot depicted the DEGs shown in B, and the dashed lines indicated log2|FC|= 1 and Padj = 0.05. The top genes identified in E are marked with green dots
To investigate the molecular signatures that affect clonal heterogeneity, especially the differentiation capacity of CMs, we identified 416 differentially expressed genes (DEGs) between the two groups. Among these, 240 genes were upregulated in PCs, whereas 176 were upregulated in NPC (log2|FC|> 1, adjusted P-value [Padj] < 0.05) (Fig. 4B). The majority of the DEGs were annotated as protein-coding genes (90%, Fig. S5B). Gene Ontology (GO) analysis revealed that PC exhibited enrichment in the development of embryonic organs, including the heart and kidney, along with DNA-binding transcription activator activity (Fig. 4C, D, and S5C). In contrast, NPC was enriched for muscle cell development, vasculogenesis, transmembrane receptor protein tyrosine kinase activity, and actin filaments. Both groups exhibited significant enrichment of the ERK1/2 signaling pathway. Furthermore, gene set enrichment analysis (GSEA) indicated the enrichment of signaling pathways regulating the pluripotency of stem cells in PC, whereas NPC was enriched for cell adhesion molecules and vascular smooth muscle contraction (Fig. S5E and F). To figure out which genes are involved in clone-specific GO terms and the ERK1/2 signaling (Figs. 3A–D, 4C, D, S4C, and D), we created a chord plot showing the relationships between DEGs and enriched pathways (Fig. 4E and F). For example, NPC showed upregulated levels of the transmembrane protein receptor tyrosine kinases TIE1 and TEK (also known as TIE2), which are key regulators of vasculature development (e.g., endothelial cells) [25, 26] and ERK1/2 activation [27]. In contrast, PC showed upregulated levels of FOXC1, a member of the FOX family involved in embryonic tissue development [28] and differentiation of early CMs [29]. These findings highlight the transcriptomic profiles between PC and NPC with varying differentiation potential for CMs.
Dysregulation of endogenous retroelements modulated nearby gene transcription in clonesEndogenous retroelements (EREs), comprising long terminal repeats (LTRs) and non-LTR transposons (Fig. 5A), constitute almost half of the human genome [30]. Previous studies have highlighted the effect of ERE expression patterns in pluripotent stem cells on various characteristics of hiPSCs, including cell morphology, pluripotency, and lineage commitment [31,32,33]. To examine the expression patterns of EREs among the clones, we mapped transcript reads onto copies of transposable elements (TEs) (Fig. 5B). The total read counts of TE contents were quantified for each clone (4.6 × 106, 4.7 × 106 and 4.7 × 106, respectively), showing consistent patterns across all clones (Fig. 5C). Differentially expressed TEs were identified between PC and NPC (log2|FC|> 1 and Padj < 0.05), with LTRs showing the highest proportions (CL1: 83.9%, CL2: 86.6%, and CL3: 84.9%). Notably, NPC exhibited an increase in the total number of significant copies of TEs when compared with PC (Fig. 5D). As copies of repeat classes are distributed across the human chromosomes [34], we compared the overall and locus-specific expression of LTRs to identify differentially expressed subtypes and their chromosomal locations. We detected 15 significant LTR subtypes (log2|FC|> 1 and Padj < 0.05) between PC and NPC. Of these, 5 subtypes (HERVK-int, HERVK11-int, LTR88c, THE1D-int, and MER52C) were highly expressed in PC, while 10 subtypes (HERVI-int, MER52C, HERV4_I-int, MER51B, LTR88b, etc.) were highly expressed in NPC (Fig. 5E). Additionally, 103 ERE loci were significantly dysregulated, with 49 of these loci belonging to LTRs from five subfamilies—ERV1 (63.3%), ERVK (2%), ERVL (8.2%), ERVL-MaLR (24.5%), and gypsy (2%) (Table S3).
Fig. 5
Assessment of variations in expression levels of endogenous retroelements (EREs). A Classification of human transposable elements (TEs). B Experimental scheme for the analysis of TEs. C Bar plot showing the proportion of total read counts of TE contents identified in RNA-seq. D Bar plot showing the proportion of differentially expressed TEs (log2|FC|> 1 and Padj < 0.05), indicating that the long terminal repeats (LTRs) showed the highest proportion. Normalized counts of the differentially expressed LTRs are presented. E Heatmap showed the expression levels of significant LTR subtypes (e.g., LTR88c, HERVI-int, and HERV4_I-int) between PC and NPC. F Heatmap displayed the expression of genes located in proximity to the identified EREs in E. The genes corresponding to DEGs are marked with *. (G-H) Loci of the genes (ANKRD2, CLEC2A, OC90, and HHLA1) were identified in the University of California, Santa Cruz (UCSC) browsers and visualized in Integrative Genomics Viewer (IGV) tracks. Two examples of dysregulated MER77 and HERVI-int integrants and expression level of the closest genes: OC90 and HHLA1 for MER77 on chromosome 8 (G), and ANKRRD2 and CLEC2A for HERVI-int on chromosome 1 and 12, respectively (H)
Given that EREs act as cis-regulatory elements to regulate the expression of neighboring genes and generate chimeric transcripts [35], we investigated differentially expressed loci near gene transcripts. We identified 21 genes adjacent to these loci (Fig. 5F and Table S4), of which 8 genes (ANKRD45, LYST, CLEC2A, RAB17, WFDC3, CEP85L, HHLA1, and OC90) are protein-coding. Among these, 5 genes (CLEC2A, RAB17, CEP85L, HHLA1, and OC90) were identified as DEGs (Fig. 4B). Consistent with previous findings [32, 34, 35], the upregulation of HHLA1 and OC90 was affected by nearby ERE loci (Fig. 5G). Unexpectedly, no reads were mapped to exons 1–4 of CLEC2A, suggesting a new transcript potentially transcribed from LTR10B1 (chr12: 10,055260–10,055,764) and HERVI-int (chr12: 10,055,800–10,059,965) (Fig. 5H and Table S4). We observed a differentially expressed ERE loci near ANKRD45, which was not identified as a DEGs (chr1: 173,607,756–173,611,588). Collectively, our analysis of EREs revealed that profiles of EREs, especially LTRs, may contribute to clonal heterogeneity by regulating nearby genes and generating new transcripts.
Clones revealed differential chromatin accessibility and susceptibility to transcription factorsThe reprogramming of hiPSCs entails global remodeling of the epigenetic state [36], where epigenetic variations, such as DNA methylation and chromatin state, play pivotal roles in the differentiation potential of hiPSCs [37]. To detect genomic chromatin accessibility between PC and NPC, we performed an ATAC-seq. An average of 9.6 × 106 fragments per sample was obtained, with a consistent distribution of peaks across genomic regions in all clones (Fig. 6A). In total, 3.1 × 105, 3.0 × 105, and 3.6 × 105 open chromatin peaks were identified in CL1, 2, and 3, respectively (Fig. 6B). PC exhibited elevated chromatin accessibility in the vicinity of transcription start site (TSS) regions (TSS ± 3 kb) compared to NPC, indicating a predisposition for active transcription (Fig. 6C). We identified 2869 differentially accessible regions (DARs) by comparing ATAC-seq peak tag counts. Among these, 2142 regions were detected in PC (log2|FC|> 0.5 and P < 0.05), whereas 727 regions were detected in NPC (Fig. 6D). Next, we examined the genomic locations of DARs, and the distal regions (> 10 kb from the TSS) accounted for 43% and 37% in PC and NPC (Fig. 6E), respectively. The promoter-proximal regions (TSS ± 3 kb) accounted for 23% and 18% of DARs in PC and NPC. Although it has been reported that the chromatin states in distal regions can affect gene expression [38, 39], we focused on how DARs in promoter-proximal regions affect gene expression status.
Fig. 6
Different chromatin statuses of human induced pluripotent stem cell (hiPSC) clones were identified using ATAC-sequencing. A Quality control of ATAC-seq. Stacked bar charts showed the distribution of peaks across genomic regions. B The circle plot of genome-wide chromatin accessibility identified in ATAC-seq. C The enriched heatmap displayed the normalized score of ATAC-seq peaks within ± 3 kb of the transcription start site (TSS). D Heatmap of differentially accessible regions (DARs, log2|FC|> 0.5 and P < 0.05) between PC and NPC. E Pie charts illustrated the distribution of DARs across genomic regions. F The volcano plot depicted the genes annotated as promoter in (E); n = 454 for PC (red) and n = 128 for NPC (blue). The dashed lines indicated log2|FC|> 0.5 and P < 0.05. G Gene Ontology (GO) analysis of the genes shown in F. H Loci of the genes (TEK and PDGFRB) were identified in University of California, Santa Cruz (UCSC) browsers and visualized in Integrative Genomics Viewer (IGV) tracks. I Heatmap displayed gene expression levels related to identified GOs (GO:1,905,772, 0007507) in G. The genes corresponding to DEGs are marked with *
We identified 454 and 128 genes with increased open chromatin around their promoters in PC and NPC, respectively (Fig. 6F). For example, chromatin accessibility (TSS ± 3 kb) of DPPA2, a key factor in resetting the epigenome to a pluripotent state during reprogramming [40], was increased in PC. We performed GO analysis to investigate the biological processes associated with the genes harboring DARs. We found that DAR-associated genes in PC were enriched in processes related to positive regulation of DNA/Nucleic acid-templated transcription (Fig. 6G), consistent with findings from our RNA-seq analysis (Fig. 4C). Despite the limited differentiation potential of NPC towards CMs, DAR-associated genes in NPC as well as PC were enriched in heart development (Fig. 6G). Specifically, 11 genes in NPC were related to heart development (GO: 0007507) (Fig. 6H and I). The expression levels of PDGFRB and TEK, which are implicated in endothelial cell differentiation [41] and atrioventricular canal development [42], were significantly upregulated in NPC. Consequently, this increased expression in NPC, driven by DARs in promoter-proximal regions, may impede CM differentiation, resulting in non-CMs with increased expression of VIM.
Next, we performed HOMER (Hypergeometric Optimization of motif EnRichment) de novo motif analysis using DARs to identify clone-specific transcription factor (TF) motifs. The ETV4 and GATA5 motifs were the top motifs in PC, whereas the TEAD2 and FOSL2 motifs were found in NPC (Fig. S6A). The ETV4-encoding ETS-domain-containing TF family plays a role in the maintenance of pluripotency in stem cells [43], and some GATA factors (GATA4, GATA5, and GATA6) are expressed in the precardiac mesoderm during heart development [44]. Although we further associated enriched motifs with the mRNA expression levels of TF families that could bind these motifs, we did not find a correlation between the motifs and the expression levels of their family members at the undifferentiated stage (Fig. S6B). Nevertheless, our results showed that the clones presented different susceptibilities to specific TFs, and they could regulate gene expression during cardiac differentiation, similar to the GATA family. Collectively, our epigenomic profile suggests that variability in the epigenetic state may influence the changes in gene expression, thereby contributing to clonal heterogeneity.
TEK and SDR42E1 were identified as marker genes for clonal heterogeneity of hiPSC-CM differentiationRecent studies have proposed reliable marker genes based on comprehensive analysis of transcriptional and epigenetic modifications [12, 13, 39]. To identify candidate genes that could predict cardiac differentiation, we ass
留言 (0)