
記住我
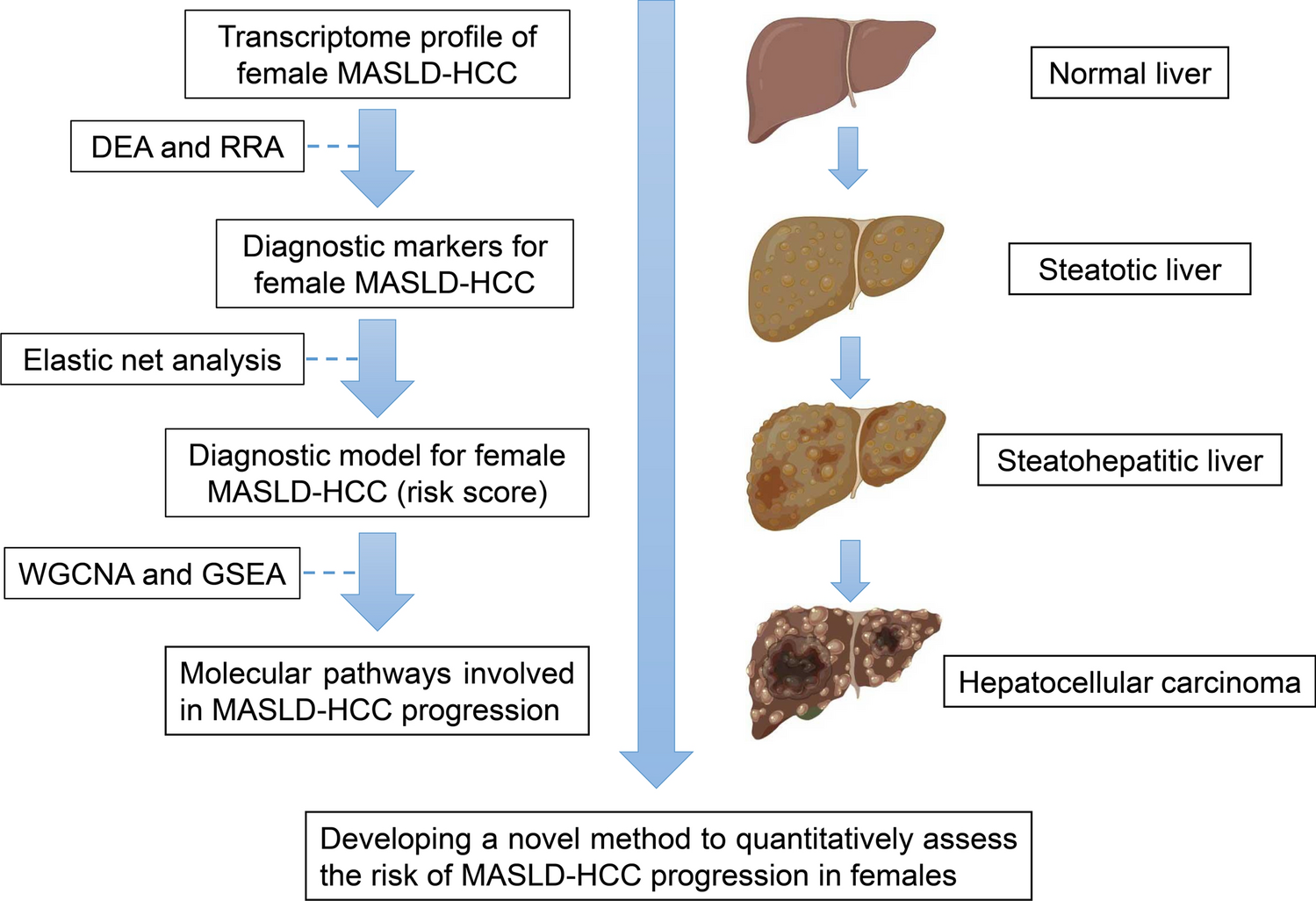





The flow chart of this study is shown in Fig. 1.
Fig. 1
Flowchart of multi-step screening strategy for bioinformatics data
3.1 Single-cell transcriptome altas of SKCMIn this study, we analyzed 4645 cells from the GSE72056 dataset, comprising melanoma, immune, and stromal cells from 19 patients [19]. The t-SNE method was used for clustering visualization, resulting in 17 distinct clusters (Figure S1). Feature expression was identified for each cluster (Figure S2A, Figure S2B), leading to the identification of seven key clusters (Fig. 2A-figure supplement 2B): B cells with high CD79A expression, NK cells with elevated NKG7 expression, melanoma cells marked by high MLANA and SOX10 expression, T cells with high CD3D expression, macrophages with elevated MARCO expression, endothelial cells with high VWF expression, and fibroblasts with elevated PDRGA expression (Fig. 2C, Figure S2C). The proportions of these clusters are shown in Fig. 2D, where T cells, melanoma cells, and NK cells are the most prevalent, while macrophages, endothelial cells, and fibroblasts are less common. Using the FindAllMarkers function, we identified the differential gene expression within each cluster.
Fig. 2
Single-cell sequencing analysis. A 7 cell types identified based on marker gene expression. B Classification of cells. A total of 7 cell types can be found, such as B cells, NK cells, Melanoma cells and T cell. C Violin plots displaying expression of marker genes for the 7 cell types across cells. D Proportional representation of different cell types. E GSVA investigation of different cell types. F Network diagram of the frequency of ligand-receptor interactions between different cell types
3.2 Enrichment analysis of single cells in scRNA-seqIn order to explore the function of differential genes in each clump, we performed GSVA enrichment analysis on seven clumps of single cells. Among them, the differential genes of B cells clusters were mainly enriched in the functions of OXIDATIVE-PHOSPHORYLATION, REACTIVE-OXYGEN-SPECIES-PATHWAY, etc. The up-regulated differential genes in Endothelial cells clusters were mainly enriched in ANGIOGENESIS, EPITHELIAL-MESENCHYMAL-TRANSITION, COAGULATION, TGF- BETA-SIGNALING, and other functions. In contrast, the down-regulated differential genes of Endothelial cells clusters were mainly enriched in the ALLOGRAFT-REJECTION function.The differential genes of Fibroblasts cells clusters were mainly enriched in the MYC-TARGETS -V1, OXIDATIVE-PHOSPHORYLATION and EPITHELIAL-MESENCHYMAL-TRANSITION functions.Macrophages Cluster differential genes were mainly enriched in INTERFERON-GAMMA-RESPONSE, TNFA-SIGNALING-VIA-NFKB INFLAMMATORY-RESPONSE and IL6-JAK-STAT3-SIGNALING.The up-regulated differential genes in NK cells clusters were mainly enriched in ALLOGRAFT -REJECTION and INTERFERON-GAMMA-RESPONSE. The down-regulated differential genes of NK cells clusters were mainly enriched in functions such as COAGULATION and OXIDATIVE-PHOSPHORYLATION. The up-regulated differential genes of Melanoma cells clumps were mainly enriched in functions such as MYC-TARGETS-V1 and OXIDATIVE-PHOSPHORYLATION. Conversely, the down-regulated differential genes of Melanoma cells clusters were mainly enriched in functions such as ALLOGRAFT-REJECTION. up-regulated differential genes of T cells clusters were mainly enriched in functions such as ALLOGRAFT-REJECTION. Conversely, the down-regulated differential genes of T cells clusters were mainly enriched for functions such as MYC-TARGETS-V1 and OXIDATIVE-PHOSPHORYLATION (Fig. 2E). From these Cluster enrichment results we found that there was a striking consistency in the pathways to which the up-regulated differential genes in the Melanoma cells clusters were enriched with the down-regulated differential genes in the T cell clusters. From this phenomenon, we hypothesised that the development of cutaneous malignant melanoma may be more related to T cells, and so we focused on the differential genes in the T cell clusters down the pathway.
These pathways, particularly MYC-TARGETS-V1 and OXIDATIVE PHOSPHORYLATION, were chosen as key upregulated functions due to their established roles in cancer biology. The MYC pathway is a critical regulator of cell proliferation, metabolism, and survival, and its dysregulation is commonly associated with tumorigenesis in many cancers, including melanoma. MYC activation drives uncontrolled cell division, contributing to melanoma aggressiveness by promoting the transcription of genes that support cell growth and division.
In parallel, OXIDATIVE PHOSPHORYLATION is crucial for energy production in rapidly proliferating tumor cells. In melanoma, metabolic reprogramming enables cancer cells to meet the increased demands for energy (ATP) and biosynthetic precursors, supporting their high proliferative rate. The upregulation of oxidative phosphorylation indicates that melanoma cells rely heavily on this metabolic process to maintain growth, especially under the often hypoxic conditions of the tumor microenvironment. Together, these pathways highlight key mechanisms that sustain melanoma malignancy and offer potential targets for therapeutic intervention.
3.3 CellChat analysisIn this study, we used "CellChat" simulation to analyse the cell–cell communication network generated by the interaction between different cells (Fig. 2F). We can see that the interaction between NK cells and macrophages is strong, Melanoma cells have strong interaction with NK cells, Fibroblasts cells, macrophages and T cells, and T cells have strong interaction with NK cells (Figure S3). Figure S4 shows that T cells were interacting strongly with Melanoma cells through the CD99-CD99 pathway. We surmised that T cells do have some relevant roles with Melanoma cells based on this result, so we extracted the T cell-related differential genes for subsequent analysis (Fig. 3A).
Fig. 3
Gene expression characterization of SKCM-infiltrating tregs and exhausted CD8 + T cells A t-SNE plot showing T lymphocyte subtypes. B Volcano plot showing differentially expressed genes in SKCM -infiltrating Tregs. C GSEA analysis results in GSE15605 D The same volcano plot as in B in the GSE46517 dataset. E GSEA analysis results in GSE46517 F The Venn graph showing the overlap of the two data sets with T cell genes
3.4 Differentially expressed genes(DEGs)Flowchart of the overall data screening strategy (Fig. 1). Two GEO datasets GSE15605 and GSE46517 were obtained for analysis. We analysed the SKCM and normal skin tissues from these two datasets for differences with the screening conditions of logFC > 0.5 and p < 0.05, and finally obtained the differential genes (Fig. 3B, D).
We performed GSEA enrichment analysis on the acquired differential genes to obtain the diseases or functions regulated by the differential genes corresponding to the two datasets.Diseases or functions enriched in the GSE15605 dataset were Allograft rejection, Antigen processing and presentation, Arginine biosynthesis, Drug metabolism—cytochrome P450, Fatty acid biosynthesis, Glycosphingolipid biosynthesis—ganglio series, Graft-versus-host disease, Terpenoid backbone biosynthesis (Fig. 3C). Diseases or functions enriched in the GSE46517 dataset were Focal adhesion, Histidine metabolism, Nicotine addiction, Ribosome, Staphylococcus aureus infection, Steroid biosynthesis, Tuberculosis, Valine, leucine and isoleucine degradation (Fig. 3E). It is predicted that the development of cutaneous malignant melanoma may be associated with changes in the above related functions.
3.5 Screening of candidate genes associated with SKCMTaking the intersection of the above modules with the obtained differential genes, the 9 genes with the highest correlation with cancer were obtained as pivotal genes (Fig. 3F).
3.6 Functional enrichment analysisTo uncover the molecular biological processes underlying common disease genes and the pathogenesis of cutaneous malignant melanoma, we conducted functional enrichment analyses on differentially expressed genes (DEGs) using GO, KEGG, and Hallmark pathways. Among the enriched pathways, oxidative phosphorylation and metabolic reprogramming were significantly highlighted, indicating a central role in melanoma progression. This aligns with findings in prostate cancer, where mitochondrial dysfunction was identified as a critical factor influencing molecular subtypes and patient prognosis [27]. These results suggest that mitochondrial dysfunction and associated metabolic changes are conserved mechanisms across different cancer types, contributing to tumor aggressiveness and treatment resistance. GO enrichment was performed with the clusterProfiler and DOSE packages in R (Fig. 4A). The analysis indicated that, in biological processes, these genes were predominantly involved in CD4-positive, alpha–beta T cell activation, and carbohydrate transmembrane transport. For cellular components, enrichment was primarily observed in tertiary granules and secretory granule membranes. In terms of molecular function, genes were mainly enriched in carbohydrate-related activities. KEGG pathway analysis revealed that common genes were primarily associated with pathways such as ubiquitin-mediated proteolysis, cell cycle, DNA replication, mismatch repair, and T cell receptor signaling (Fig. 4B). Hallmark gene set enrichment analysis showed concentration in pathways linked to Myc targets v1, protein secretion, G2/M checkpoint, mitotic spindle, E2F targets, unfolded protein response, PI3K/AKT/mTOR signaling, oxidative phosphorylation, heme metabolism, MTORC1 signaling, and DNA repair (Fig. 4C). The results from GO, KEGG, and Hallmark analyses were consistent with existing reports on cutaneous malignant melanoma, indicating their reliability.
Fig. 4
Enrichment analysis and machine learning. A Based on GO enrichment analysis, these genes are mainly involved in CD4-positive, alpha–beta T cell activation and carbohydrate transmembrane transport in biological processes, and in terms of cellular components are mainly focused on tertiary granule and secretory granule membranes, while in terms of molecular functions, they mainly exhibit carbohydrate transmembrane transport activity. B KEGG-enriched analyzed genes were mainly focused on pathways related to Ubiquitin mediated proteolysis, Cell cycle, and DNA replication. C Enrichment analysis of the Hallmark gene set showed that common genes were mainly concentrated in pathways related to Myc targets v1, Protein secretion, G2/M checkpoint, etc. D Machine learning model comparison using 7 different machine learning algorithms. E Receiver operating characteristics (ROC) curves for multiple machine learning models. F The “logreg” machine learning algorithm model exhibits the highest AUC of 0.995
3.7 Machine learningTo elucidate the progression of cutaneous malignant melanoma, we utilized the identified hub genes to construct predictive models for further examination. The GSE15605 dataset served as the training cohort, while the GSE46517 dataset was used to evaluate the models' predictive capabilities. We applied seven distinct machine learning algorithms, optimizing each model's parameters through five repetitions of ten-fold cross-validation (Fig. 4D). The models' performance was assessed by examining their area under the curve (AUC) values in the validation cohort (Fig. 4E). Through these stringent evaluations, the "logreg" machine learning algorithm emerged as the top performer, achieving the highest AUC of 0.995 (Fig. 4F). Additionally, we developed a linear regression model to assess the pathogenicity of the nine hub genes for cutaneous malignant melanoma. Using a lasso regression algorithm, we identified three key genes—PIP4K2A, SLC2A3, and RORA—that were strongly linked to disease pathogenicity (Fig. 5A, B). Among these, PIP4K2A had the highest weight in the lasso regression model, warranting a more detailed subsequent analysis.
Fig. 5
Identification of key genes strongly associated with disease pathogenicity and survival analysis. A Binomial deviation of overall survival (OS) for LASSO coefficient curve. B LASSO coefficient profiles of genes. C Patients in the PIP4K2A high-expression group in cutaneous malignant melanoma have a somewhat shorter survival time. D Meta-survival analysis results of PIP4K2A expression. E Higher expression of PIP4K2A in elderly patients > 65 years of age. F Higher expression of PIP4K2A in patients with clinical manifestations of ulceration. G, H PIP4K2A is more highly expressed in cutaneous malignant melanoma tissue samples. I Higher PIP4K2A expression at higher T stage. J The expression of PIP4K2A showed differences in the performance of individual staging
3.8 Survival analysisBased on the transcriptomic data of GSE190113 with matched clinical data, the Kaplan–Meier survival curves indicated that the survival time of patients in the group with high expression of PIP4K2A in cutaneous malignant melanoma was a little shorter (Fig. 5C). In the results of Meta-survival analysis, the HR was 1.21 for the Fixed effect model and 1.26 for the Random effects model, further testifying that PIP4K2A is a risky prognostic factor in cutaneous malignant melanoma (Fig. 5D). Clinical data according to the GSE22153 dataset showed that PIP4K2A also exhibited a significant role in cutaneous malignant melanoma in the elderly, with higher expression of PIP4K2A in elderly patients greater than 65 years of age compared to those under 65 years of age (Fig. 5E). According to clinical data from the GSE98394 dataset, PIP4K2A expression was higher in patients with cutaneous malignant melanoma with clinical manifestations of ulceration (Fig. 5F). On the tissue side, both the GSE98394 and GSE46517 datasets showed higher expression of PIP4K2A in cutaneous malignant melanoma tissue samples (Fig. 5G, H). According to the clinical data of GSE98394, the higher the T-stage the higher the expression of PIP4K2A in cutaneous malignant melanoma was correspondingly higher (Fig. 5I), demonstrating an enhanced proliferative capacity of cutaneous malignant melanoma with high PIP4K2A expression. In terms of cancer stage, clinical data from the GSE98394 datasets showed differences in the expression of PIP4K2A across stages (Fig. 5J).
3.9 Immunoinfiltration analysisUtilizing preprocessing from the BEST online platform, we analyzed datasets GSE22153, GSE133713, GSE98394, GSE65904, TCGA_SKCM, GSE46517, GSE54467, GSE53118, GSE99898, GSE22154, GSE78220, GSE100797, and GSE190113. Immune infiltration in cutaneous malignant melanoma, particularly in the high PIP4K2A expression group, was evaluated using the CIBERSORT, CIBERSORT_ABS, EPIC, ESTIMATE, MCPcounter, Quantiseq, TIMER, and xCell algorithms (Fig. 6). In the CIBERSORT_ABS analysis, significant infiltration was observed in follicular helper T (Tfh) cells, naive B cells, activated memory CD4 + T cells, activated dendritic cells, and resting memory CD4 + T cells. The Quantiseq algorithm highlighted notable infiltration of B cells and dendritic cells. According to the CIBERSORT results, naive B cells, Tfh cells, activated memory CD4 + T cells, M1 macrophages, resting memory CD4 + T cells, and activated dendritic cells showed pronounced infiltration. The xCell algorithm revealed high levels of B cells, naive B cells, mast cells, naive CD4 + T cells, memory B cells, class-switched memory B cells, memory CD4 + T cells, CD4 + T cells, plasmacytoid dendritic cells (pDC), central memory CD8 + T cells (CD8 + TCM), CD8 + T cells, activated dendritic cells (aDC), conventional dendritic cells (cDC), NK cells, effector memory CD8 + T cells (CD8 + TEM), Tregs, granulocyte-monocyte progenitors (GMP), gamma delta T cells (Tgd), macrophages, M1 macrophages, and melanocytes. MCPcounter analysis showed significant infiltration of T cells, B lineage cells, myeloid dendritic cells, CD8 + T cells, monocytic lineage cells, cytotoxic lymphocytes, and NK cells. TIMER analysis indicated higher levels of infiltration in B cells, CD8 + T cells, dendritic cells, CD4 + T cells, neutrophils, and macrophages.
Fig. 6
Immunoinfiltration analysis of PIP4K2A expression
3.10 Predictive analysis of drug therapyIn the drug therapy prediction analysis, in the PRISM database, we found that high PIP4K2A expressing cutaneous malignant melanomas were resistant to carbimazole, methimazole, barnidipine, talniflumate, cyanocobalamin, sulisobenzone, dexniguldipine, brucine, proxyphylline, AGI-6780, lomefloxacin, hydroflumethiazide, salazodine, bicalutamide, bemesetron, uric -acid, otenzepad, fenobam, ufenamate and pirodavir, among a range of other drugs, produced resistance. In the CTRP database analysis we found that in the high PIP4K2A expression group for erismodegib, BRD-K64610608, Platin, BRD-K16147474, semagacestat, AGK-2, BRD-K09587429, SB-431542, austocystin D, etomoxir, spautin-1, erlotinib, canertinib, MK- 0752, BRD-K17060750, CI-976, CAY10576, hyperforin, procarbazine and silmitasertib, and a host of other drugs were resistant. In the GDSC1 database analysis we found that high PIP4K2A expressing cutaneous malignant melanoma was resistant to AST-1306_381, JNJ38877605_284, Tanespimycin_1026, AZD8931_1416, Dyrk1b_0191_1407, Pilaralisib_372, Trametinib_1372, Selumetinib_1498, Refametinib_1526, Refametinib_1014 and Cetuximab_1114, amongst a range of other drugs, have produced resistance. In the GDSC2 database we found that cutaneous malignant melanoma in the high PIP4K2A expression group was resistant to VX-11e_2096, SCH772984_1564, Trametinib_1372, Selumetinib_1736, PD0325901_1060, Sapitinib_1549, ERK_6604_1714 and ERK_2440_1713, and a range of other drugs produced resistance. These data assess that high PIP4K2A expression is indeed a risk factor for cutaneous malignant melanoma drug therapy. (Figure S5).
3.11 Immunotherapy predictionWe plotted AUC curves based on the Homet cohort, Cho cohort, Kim cohort, and Nathanson cohort, and verified that the high-expression PIP4K2A group had a poor response to Anti-PD-1, Anti-PD-L1 and Anti-CTLA-4 immunotherapy were all poorly responsive (Fig. 7A–D).
Fig. 7
Immunotherapy predictive analytics and immunohistochemistry. A–D Prediction of immunotherapy sensitivity in different cohorts. (E)The expression of PIP4K2A was significantly higher in cutaneous melanoma tissues than in normal skin tissues
3.12 Expression of PIP4K2A in cutaneous melanomaTo explore the significance of PIP4K2A, immunohistochemical experiments were performed using four normal skin tissues as a control group and four cutaneous melanoma tissues as an experimental group to assess the expression of PIP4K2A in both tissue types (Fig. 7E). The immunohistochemical staining results showed that the expression of PIP4K2A was significantly higher in cutaneous melanoma tissues compared to normal skin tissues. In cutaneous melanoma tissues, PIP4K2A was strongly positively stained and widely distributed with high staining intensity, predominantly localized in the tumor cells. These tumor cells were characterized by irregular shapes and dense, disorganized cellular structures, indicating that PIP4K2A plays a potential role in promoting melanoma cell proliferation and tumor progression.
In contrast, in normal skin tissues, PIP4K2A expression was lower, with lighter staining intensity primarily observed in the basal layer of the epidermis. This layer contains stem-like keratinocytes that are responsible for skin regeneration and repair, suggesting that PIP4K2A in normal skin is involved in regulating basic cellular growth and maintenance functions. No significant PIP4K2A expression was observed in other cell types such as immune or stromal cells in the normal skin samples.
These results are consistent with our single-cell transcriptome and machine learning analyses, further confirming that PIP4K2A is an important malignant prognostic marker for cutaneous melanoma. The differential expression between melanoma and normal skin highlights PIP4K2A's role in the aggressiveness of melanoma, while its restricted expression in normal skin suggests a more regulated involvement in normal tissue homeostasis.
留言 (0)