Cell culture
Patient-derived GBM stem cells (448, 83, X01) [27] and (528, 022) [49] were cultured in DMEM/F-12 medium, enhanced with B27 supplement (Invitrogen), and further augmented with EGF (10 ng/ml, R&D Systems) and bFGF (5 ng/ml, R&D Systems). Meanwhile, 293T cells were maintained in DMEM medium (Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich). CCF642, securinine, and V-ZAD-FMK were purchased from TOPSCIENCE. V-ZAD-FMK, Z-DEVE-FMK dimethyl fumarate, ferrostatin-1, and the recombinant P4HB protein were purchased from MedChemExpress. To ensure the integrity of the cultures, all cells were routinely screened for mycoplasma contamination. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Bioinformatics analysis
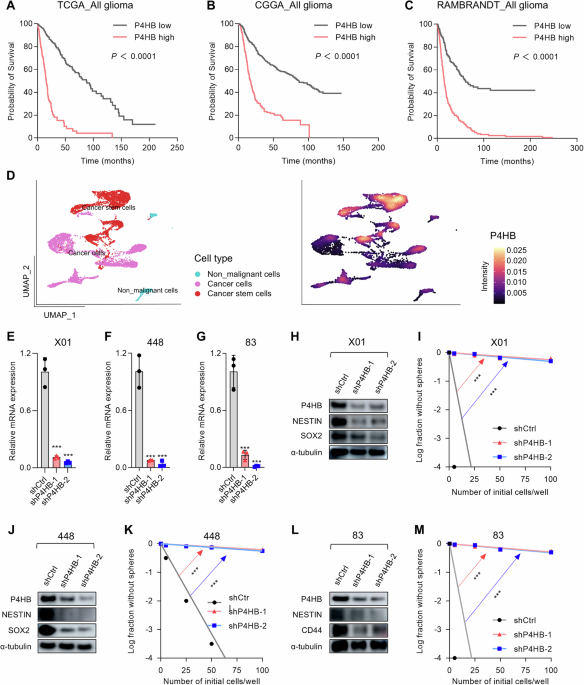
Expression matrices for P4HB in glioma patient samples were sourced from the GlioVis data portal (http://gliovis.bioinfo.cnio.es/) [50], which aggregates processed datasets from the Cancer Genome Atlas (TCGA), Chinese Glioma Genome Atlas (CGGA), and Repository of Molecular Brain Neoplasia Data (REMBRANDT) and CPTAC (The Clinical Proteomic Tumor Analysis Consortium). Detailed methodologies for data processing are available on the GlioVis portal help page. Kaplan–Meier survival curves were constructed across each dataset to compare overall survival probabilities between groups characterized by high and low expression levels of P4HB. The statistical significance of differences in survival outcomes was evaluated using log-rank and Wilcoxon tests. The expression plots, gene correlation plots, and survival curves were visualized by GraphPad Prism (v8.0).
To further investigate the specific expression of P4HB in GBM cells and GBM stem cells in GBM, single-cell RNA sequencing (scRNA-seq) data from human GBM samples were analyzed. This dataset was acquired from the GEO data portal and processed using R software (v4.1.3). The analysis workflow commenced with the loading of raw counts using the ‘Read10X()’ function in the SEURAT package (v4.3.0) [51], followed by the generation of a Seurat object. Data normalization was performed with ‘NormalizeData()’, and variable features were identified using ‘FindVariableFeatures()’. Subsequent data scaling was accomplished with ‘ScaleData()’. Principal Component Analysis (PCA) was implemented via ‘RunPCA’, with visualization conducted using ‘RunUMAP’. Marker genes were identified with ‘FindMarkers()’, and cellular clusters were annotated. The expression of the P4HB gene was distinctly highlighted on the 2D UMAP plot to delineate its distribution among various cellular subpopulations.
Lentivirus construction and infection
The shRNA-expressing lentiviral constructs targeting P4HB were engineered by ligating annealed oligomers into the PLKO.1 puro vector (Addgene). The sequences of the P4HB shRNA oligos are listed in Supplementary Table 1. All constructs were confirmed via DNA sequencing (Tsingke Biotech). For lentivirus production, 293T cells (3 × 106) were seeded in 100-mm culture dishes and incubated for 24 h. Subsequently, the cells were co-transfected with 4.5 μg of target plasmids and 4.5 μg of helper plasmids (psPAX2, pMD2.G, Addgene) using 27 μl of Lipofectamine 2000 (Invitrogen). The culture medium was replaced 6 h post-transfection. After 48 h, the medium containing the lentiviral particles was collected. The viral particles were then concentrated and purified using a PEG8000 concentrator (Sangon Biotech). For infection, GBM stem cells were exposed to lentivirus in the presence of 10 μg/ml polybrene (Sigma-Aldrich) to enhance viral entry.
Quantitative RT-PCR
Total RNA was extracted from GSCs using the FastPure® Cell/Tissue Total RNA Isolation Kit (Vazyme). Subsequently, 1 μg of RNA was reverse transcribed into cDNA utilizing the HiScript II One Step RT-PCR Kit (Vazyme). Quantitative RT-PCR analysis was conducted on a LightCycler 480 II real-time PCR system (Roche) employing the HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme). The expression levels of the target genes were quantified relative to the housekeeping gene GAPDH. Primer sequences are provided in the Supplementary information primer list.
Westernblot analysis
Protein samples were extracted from GSCs using RIPA buffer supplemented with complete protease inhibitors (Solarbio, R0010), followed by centrifugation at 13,000 rpm for 15 min at 4 °C. The protein samples were subjected to electrophoresis analysis, transferred to PVDF membranes (Millipore), and blocked with 5% skim milk (Vazyme). For the inhibitor study, 1 μM of CCF642 was added to the cell culture medium the day after seeding. The cells were harvested at subsequent time points: 0, 2, 3, 6, 12, 24, and 48 h. In the nuclear-cytoplasmic separation experiment, nuclear and cytoplasmic proteins were isolated using a nuclear-cytoplasmic separation kit (Beyotime), according to the manufacturer’s instructions. Western blots were incubated with primary antibodies targeting P4HB (Proteintech, #D154013), α-Tubulin (Proteintech, #11224-1-AP), GAPDH (HUABIO, #EM1101), Vinculin (Proteintech, #26520-1-AP), Caspase-3 and Cleaved Caspase-3 (CC3, Cell Signaling Technology, #14220, #9664), PARP and Cleaved PARP (C-PARP, Cell Signaling Technology, #9542, #5625), BCL-2 (1:1000, Abcam, #ERP17509), β-catenin (Beyotime, #AC106), LRP6 (Cell Signaling Technology, #2560T), P-LRP6 (ZENBIO, R30284), Cyclin D1 (Proteintech, #60186-1-Ig), NESTIN (Thermo Fisher Scientific, #PA5-11887), SOX2 (R&D Systems, #AF2018-SP), CD44 (R&D Systems, #BBA10), and Lamin B1 (Proteintech, #12987-1-AP) overnight at 4 °C. The recombinant protein was purchased from MCE (HY-P71917). The immunoreactive bands were visualized using peroxidase-conjugated secondary antibodies and detected with the Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare).
Limit-dilution assay
GSCs were incubated in 96-well plates containing DMEM/F-12 supplemented with B27, 10 ng/ml of EGF, and 5 ng/ml of bFGF at decreasing numbers (100, 50, 25, and 5 cells/well), and the tumorsphere-forming ability was determined by counting wells without tumorspheres after incubation for 1 week. For the inhibitor study, 0.1 μM of CCF642 was added to the cell culture medium the following day after seeding. The results were evaluated using the Extreme Limiting Dilution Analysis function (ELDA, http://bioinf.wehi.edu.au/software/elda/).
Cell proliferation assay
GSCs were seeded into 96-well plates at a density of 1000 cells per well to assess their proliferation across various culture conditions. This study was conducted over a duration of either 24 h or 6 days, with assessments carried out on days 0, 2, 4, and 6. Cell proliferation was quantified using the Cell Counting Kit-8 (CCK-8) assay every two days. Absorbance OD values were obtained using a Multiskan molecular device (Thermo Scientific) following the manufacturer’s protocols. Cell viability was determined by measuring the absorbance at 450 nm of the culture medium. The result was analyzed and visualized using GraphPad Prism (v8.0).
Cell apoptosis assay
To evaluate apoptosis in glioblastoma stem cells (GSCs) transfected with P4HB shRNA, the cells were plated at a density of 5 × 105 cells per well in 6-well plates. Two days post-transfection, GSCs were collected and dual-stained with Annexin-V and propidium iodide (PI) in the binding buffer. Apoptotic rates were quantified using flow cytometry (BD Biosciences). To assess apoptosis induced by the P4HB inhibitor CCF642, GSCs were seeded in 6-well plates and incubated overnight. Subsequently, various concentrations of CCF642 were administered, and the cells were incubated for an additional 24 h. Apoptosis was determined using the aforementioned staining and flow cytometry techniques. Data were analyzed using FlowJo software.
Mice model
All animal experiments were conducted in accordance with protocols approved by the Animal Care and Use Committee of the Laboratory Animal Center at Henan University, China (HUSOM2023-462). Mice were group-housed in ventilated cages under controlled temperature and humidity, with a 12-h light-dark cycle. Each animal was randomized by body weight before the experiments. For the orthotopic mouse model, 1 × 104 luciferin-labeling 83 shCtrl or shP4HB GSCs were first resuspended and then transplanted into the left striatum of 5-week-old female BALB/c nude mice via stereotactic injection. The injection coordinates were 2.2 mm to the left of the midline, 0.2 mm posterior to the bregma, and at a depth of 3.5 mm. In a similar procedure for the treatment assay, 1 × 105 X01 GSCs were transplanted into the mouse brain. After 10 days, the mice were treated with a control reagent (PBS) or 15 mg/kg of securinine via intraperitoneal (I.P.) injection, 5 days a week for 3 consecutive weeks. After the injection, the mice were anesthetized and their tumor luminescence intensity was monitored by the Lumina IVIS III system, the body weight of the mice was individually measured every 2 days. The mice were euthanized using CO2 by the animal center when a 20% reduction in body weight or severe neurological symptoms were observed, the brain of each mouse was harvested and fixed in 4% paraformaldehyde for 24 h at 4 °C for subsequent analysis like Hematoxylin and Eosin (H&E) staining. To evaluate the safety and biocompatibility of securinine, a comprehensive study was conducted using healthy BALB/c female mice aged 6–8 weeks. Following 1-week securinine administration (15 mg/kg, 5 times, I.P.), routine hematological parameters were analyzed, including white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts, along with blood chemistry markers such as aspartate aminotransferas (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), albumin (ALB), urea (UREA), creatinine (CREA), and uric acid (UA). In addition, core pro-inflammatory cytokines (Il-1β, Il-6, and Tnf-α) were measured in the liver and kidney tissues to assess potential inflammatory responses. Histological analyses were performed to examine any necrosis or apoptosis in major organs. Survival data were analyzed using GraphPad Prism (v8.0).
Hematoxylin and Eosin (H&E) stain
After fixation, the brains were processed through a series of alcohols, cleared in xylene, and embedded in paraffin. Sections were cut at 4 µm using a microtome and mounted on glass slides. For H&E staining, sections were deparaffinized in xylene, rehydrated through graded ethanol to water, and stained with hematoxylin to highlight nuclei. After rinsing and differentiating in acid alcohol, the sections were blued in alkaline water and counterstained with eosin to highlight cytoplasmic components. The slides were then dehydrated, cleared in xylene, and then coverslipped. Histological evaluation was performed under a light microscope to assess tissue architecture and cellular morphology.
RNA-seq analysis
Total RNA was extracted from X01 shCtrl and shP4HB GSCs using the TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. RNA sequencing libraries were prepared from 1 µg of total RNA using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB Biolabs), following the manufacturer’s protocol. Briefly, mRNA was purified and fragmented, followed by cDNA synthesis, end repair, A-tailing, adapter ligation, and PCR enrichment. Library quality was assessed on an Agilent 2100 Bioanalyzer, and quantification was performed using the Qubit DNA HS Assay Kit (Invitrogen). Sequencing was performed on an Illumina NovaSeq 6000 platform, using paired-end reads. Raw sequencing reads were first subjected to quality control using FastQC (Babraham Bioinformatics) to assess base quality and sequence contamination. After cleaning, reads were aligned to the human reference genome (GRCh38) using the STAR RNA-seq alignment tool (v2.7). Default parameters were used, with adjustments for known splice junctions to improve alignment accuracy. Differential expression analysis was conducted using the DESeq2 (v1.34.0) R package. Genes with an adjusted P-value < 0.05 and a log2 fold change greater than 1.5 were considered significantly differentially expressed.
Enzyme-linked immunosorbent assay
Serum samples from glioma patients and non-patients were collected from Henan Provincial People’s Hospital and Chinese PLA General Hospital. Blood samples were centrifuged at 3500 rpm for 5 min at room temperature, and serum aliquots were subsequently stored at −80 °C. P4HB concentrations in both patient and non-patient serum samples were determined using a Human Protein Disulfide Isomerase (PDI) ELISA Kit (CUSABIO), following the manufacturer’s protocol. This study was approved by the Ethics Committees of Henan Provincial People’s Hospital (Approval No: 2020-107) and Chinese PLA General Hospital (Approval No: S2021-095-02), and all participants provided informed consent prior to sample collection.
Statistics and reproducibility
All data are presented as means ± SD from three independent experiments. Survival curves were plotted using the Kaplan–Meier method. Multiple datasets were compared using ANOVA, followed by the log-rank test for survival analysis. Two-group comparisons were made using a two-tailed Student’s t-test. P values < 0.05 were considered statistically significant. Each experiment was performed in triplicate.







留言 (0)