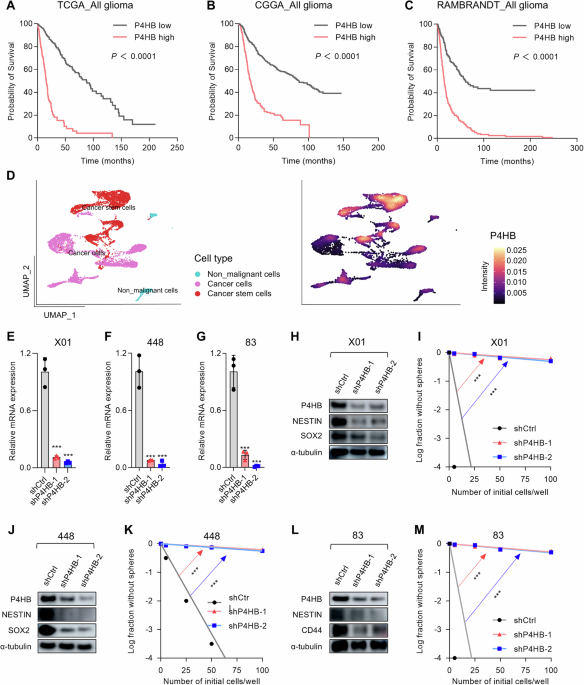
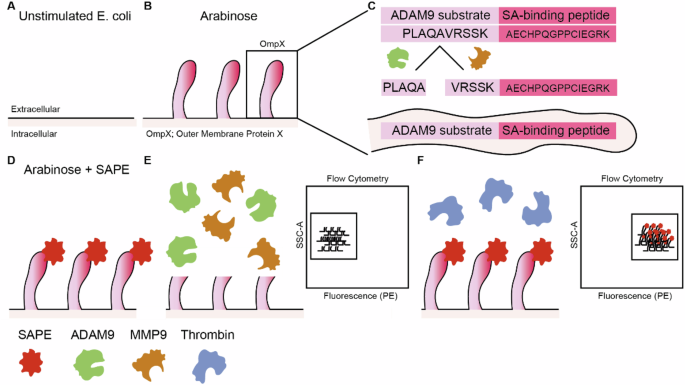
Generation ADAM9-responsive E. coli and CLiPS Library
To construct the pB33eCPX-ADAM9-SAbp plasmid, we introduced a streptavidin-binding peptide (SAbp) with the amino acid sequence AECHPQGPPCIEGRK [57] fused to an ADAM9 cleavage sequence (ADAM9seq) with the amino acid sequence PLAQAVRSSK [38] into pB33eCPX (Addgene #23336; AddGene, Watertown, MA, USA [58]). To this end, the KpnI-HindIII fragment of pB33eCPX was replaced by a GeneArt Strings DNA fragment (Thermo Fisher Scientific, Waltham, MA, USA) consisting of a modified KpnI-HindIII fragment containing the GCAGAATGCCATCCGCAGGGCCCGCCGTGCATTGAAGGTCGTAAAGCGGCGCCGCTGGCGCAGGCGGTGCGCAGCAGCAAA SAbp-ADAM9seq coding sequence (with the streptavidin-binding peptide in underscore, a short spacer in bold, and the ADAM9-responsive cleavage site in italic) immediately downstream of the ggccagtctggccag sequence coding for the flexible GQSGQ linker adjacent to the OmpX signal peptide. To generate the CLiPS Library containing six random amino acids fused to SAbp (pB33eCPX-random-SAbp), the ADAM9seq from pB33eCPX-ADAM9-SAbp was replaced by 6 NNK codons potentially coding for all amino acids and the amber stop codon using a pool of GeneArt Strings DNA fragments. pB33eCPX-ADAM9-SAbp and pB33eCPX-random-SAbp plasmids were electroporated into electro-competent TOP10F yielding 1.4 × 107 transformants that were collected in LB broth supplemented with 25% glycerol before storage at −80 °C in a concentration of approximately 1 × 108 bacteria per µl. For the remainder of the paper, the ADAM9-responsive bacteria will be referred to as ADAM9 bacteria.
Validation of ADAM9-responsive bacteria and CLiPS Library
Successful generation of ADAM9 bacteria and the CLiPS Library was confirmed by BigDye Terminator analysis. Plasmid DNA was isolated using the NucleoBond Xtra Midi kit (BIOKE, Leiden, The Netherlands) following the manufacturer’s protocol. The precipitated plasmid DNA was washed twice by adding 70% ethanol and centrifugation at 14,000 rpm for 15 min at RT. After air-drying, the DNA was dissolved in 100 µL RNase-free water and stored at −20 °C. Next, 1 µg plasmid DNA was used in BigDye terminator 3.1 (Applied Biosystems, Thermo Fisher Scientific) Sanger sequencing reaction using the forward primer 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTTCTGGCTTTCACCGCAG-3’.
Bacterial labeling optimization
To optimize the labeling and cleavage protocol, ADAM9 bacteria were grown overnight in 5 mL LB. The following morning, this culture was diluted 100-fold and grown for 1 h at 37 °C after which the bacteria were induced with varying concentrations of arabinose for various time points. Next, the bacteria were reconstituted to 109 bacteria/mL, and 104–107 bacteria were subsequently incubated with streptavidin bound R-phycoerythrin (SAPE; 1.25–20 mg/mL, Thermo Fisher Scientific) for either 30 min, 1 or 2 h under static conditions at room temperature. Finally, the bacteria were washed twice with ice-cold PBS, and the SAPE signal was measured by BD FACSCanto II (BD Biosciences, Franklin Lakes, NJ, USA) at 585 nM. Similar experiments were performed after the generation of the CLiPS Library. All generated data were analyzed using FLOWJO v10 (FlowJo LLC). The gating strategy to discriminate SAPE-positive bacteria from SAPE-negative bacteria is shown in Supplemental Fig. 10.
Cell culture
Human PANC-1, BxPC-3, MIA PaCa-2, Capan-1, and Capan-2 cells (all ATCC, Manassas, VA, USA) were grown in DMEM (Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum, 100 units/mL penicillin and 500 µg/mL streptomycin (all Lonza). THP-1 cells (ATCC) were cultured in RPMI (Lonza) supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 500 µg/mL streptomycin. Cells were cultured in a humidified incubator at 37 °C and 5% CO2. Monthly mycoplasma tests were performed on all cell lines, and their identity was confirmed yearly by STR profiling.
Preparation lysates
For CLiPS experiments, 2.5 × 106 PANC-1 (ATCC, Manassas, VA, USA) or 2.5 × 106 whole blood cells were used for lysate preparation. Whole blood was collected from a single healthy volunteer (following institutional standard operating protocol and under the approval of the Medical Ethics Review Committee) in lithium-heparin-coated blood tubes (BD, Franklin Lakes, NJ, USA). Next, cells were pelleted by centrifugation at 800 rpm for 3 min, the supernatant was discarded, and cells were resuspended in 1 mL ice-cold PBS. Cells (PDAC or the resuspended blood cells) were lysed on ice by needle sonication using a Vibra-Cell X-130 (Sonic & Materials Inc, CT, Newtown, USA) with as protocol 5 rounds of 5 s pulses at 30% amplitude followed by 5 s of pause. To remove remaining cell debris, the lysed samples were spun down at 600 rpm for 3 min, and the supernatant was aliquoted in 200 µL portions and stored at −20 °C. For FRET-peptide cleavage experiments, 1 × 106 cells/mL were lysed as described above. Freshly excised murine lung, liver, kidney, and colon were freeze-dried using liquid nitrogen, subsequently frozen using dry ice, and homogenized in 300 µL PBS using TissueLyser LT (Qiagen, Venlo, The Netherlands) at 50 Hz for 2 minutes. Next, homogenized tissues were centrifuged at 800 rpm for 3 min, the supernatant was transferred to a new tube, and the protein concentration was measured using the Nanodrop 2000 (Thermo Fisher Scientific).
Cleavage assays of ADAM9 bacteria and CLiPS library
Overnight cultures of ADAM9- or CLiPS Library bacteria were subcultured by dilution into fresh LB (1:100) and subsequently grown for 45 min at 37 °C before adding 0.04% arabinose. After 1 hour of arabinose induction, bacteria were labeled with a final concentration of 20 mg/mL SAPE for 1 h. Next, 107 bacteria resuspended in 10 µl PBS were incubated with either 10 µL PBS, reaction buffer (50 mM Tris-HCl, 20 mM NaCl, 2 mM CaCl2 and 10 µM ZnCl2 in PBS), or a final concentration of 75 nM thrombin (Sigma-Aldrich, St. Louis, MO, USA), 10 nM rADAM9 (R&D Systems, Minneapolis, MN, USA), 200 nM rMMP9 (Sigma-Aldrich), and 200 nM rMMP9 + 1 µM S3304 or dilutions of PANC-1 and THP-1 lysates. After 3 h, the bacteria were washed twice with PBS, resuspended in 100 µL PBS, and analyzed on a FACS Canto II using unlabeled bacteria as negative control. All generated data were analyzed using FLOWJO v10 (FlowJo LLC). The gating strategy to discriminate SAPE-positive bacteria from SAPE-negative bacteria is shown in Supplemental Fig. 11.
CLiPS library sorting experiments and culture processing
As we aimed to include the complexity of the CLiPS Library tenfold, we performed the sorting experiments with 1 × 108 bacteria. This constitutes a ten-fold increase of bacteria to label compared to the cleavage experiments to validate the CLiPS protocol, therefore we performed one more round of labeling optimization. Hereto, we found that SAPE-labeling did not improve when increasing the amount of SAPE (Supplemental Fig. 1F). However, it prompted us to increase the amount of arabinose to 0.12% and to extend the incubation period to 2 h (Supplemental Fig. 1G, H). To streamline the sorting process, we first assessed the impact of sorting speed, sheath fluid, and centrifugation on experimental success. Our findings indicate that increasing the sorting speed and shortening the sorting procedure, thereby reducing the exposure time of sorted bacteria to sheath fluid (i.e., sorting fluid), did not affect sorting efficiency (Supplemental Fig. 1I). As sorting takes multiple hours, the incubation time in sheath fluid differs among sorted bacteria; however, we observed no detrimental effects on cell viability across different exposure times to sheath fluid (Supplemental Fig. 1J). Sorting a high number of bacteria, up to 100 million, yields a substantial volume of bacteria in sheath fluid, ideally minimized to facilitate outgrowth in smaller bacteria cultures. Importantly, we observed a near-complete loss of bacterial outgrowth when centrifuging the collected bacteria in sheath fluid prior to initiating a new culture (Supplemental Fig. 4K). To obviate the need for centrifugation post-sorting, we explored the dilution of sheath fluid in an LB medium to support bacterial outgrowth (Supplemental Fig. 4L). To eliminate substrates from the CLiPS Library that express incorrect substrate and binding peptides (e.g., stop codons or sequences containing frame-shift mutations) the labeled CLiPS Library was first sorted twice without any incubation after which SAPE positive bacteria were selected, the resulting cultures are termed Enrich-1 and Enrich-2. Unlabeled bacteria were used to set the positive sorting gates. Next, the CLiPS Library resulting from Enrich-2 was incubated with 200 µL lysates from PANC-1 or human whole blood cells using a sequential incubation series as shown in Fig. 3. All sorting rounds were performed on a SH800 Cell Sorter (Sony Biotechnology, San Jose, CA, USA) using a 70 µm chip at 8000–10,000 events per second. Before sorting, the SH800 and sorting chip were calibrated using Sony SH800 setup beads (Sony Biotechnology). All generated data were analyzed using FLOWJO v10 (FlowJo LLC) similar as described for the protocol optimization experiments. After sorting, 10 µL of the collected SAPE-positive bacteria were plated to estimate recovery rates, and the remaining bacteria were grown overnight in 1 L LB containing 20 µg/mL chloramphenicol. The next day, 200 mL overnight culture was pelleted by centrifugation at 3000×g for 10 min at 4 °C. Dried pellets were stored at −20 °C, and plasmid DNA was isolated as described above. At the same time, 400 mL overnight culture was pelleted, supernatant discarded, and bacteria were resuspended in 10 mL LB and stored as 50:50 glycerol stock. 10 µL of the resuspended bacteria were used to determine the number of bacteria per µL stock by plating.
Sample preparation and next-generation sequencing
Amplification of the region of interest (i.e., the 18 base pairs coding for the random amino acids) and incorporation of overhang adapters was carried out in a 20 µl reaction volume consisting of 5 ng plasmid DNA, 12.5 µl HotStart Ready Mix (Roche, Woerden, the Netherlands), 2 µl forward and reverse primers (10 µM; see Table 4), and 6.5 µl H2O. Samples were subjected to 3 min 95 °C, followed by 25 cycles of 30 s 95 °C, 30 s 55 °C, 30 s 72 °C, followed by 5 min 72 °C. PCR products were cleaned using AMPure XP beads (Beckman Coulter, Brea, CA, USA) following the manufacturer’s protocol. Next, the cleaned PCR products were barcoded by index PCR. 5 µl PCR product, 1 µl forward and reverse index primers (10 µM; see Table 4), 25 µl KAPA HiFi HotStart ReadyMix and 10 µl H2O were subjected to 3 min 95 °C, followed by 8 cycles of 30 s 95 °C, 30 s 55 °C, 30 s 72 °C, followed by 5 min 72 °C. PCR products were cleaned using AMPure XP beads. The concentrations of the final PCR products were determined using D5000 DNA ScreenTape (Agilent, Santa Clara, CA, USA), measured on a Tapestation 4200 (Agilent), and finally, equal amounts per sample were pooled to a final concentration of 10 nM. The resulting library pool was sequenced on a NovaSeq 6000 (Illumina, San Diego, CA, USA) with a depth of 700 M reads (i.e., 100 M/sample).
Table 4 Primers used for BigDye Terminator (BDT) and sample preparation for Next Generation Sequencing.Bioinformatics analysis
Raw DNA sequencing reads were separated into matching samples using index barcoding and analyzed using custom Perl scripts to determine the total number of DNA sequences, total number of DNA sequences matching linker sequences, and total number of uniquely matched DNA sequences per sample. The variable DNA region was translated into the corresponding peptide sequence. Subsequently, the number of peptide sequences and the number of uniquely matched peptide sequences were determined. Given the observed differences in sequencing depth between samples we decided to calculate the ratio (i.e., abundance) of every individual peptide based on the total amount of reads per sample, allowing us to directly compare samples. Next, the fold change of every individual unique peptide sequence as the experiment progressed was calculated by comparing the ratio in a given sample (PDAC-1, PDAC-2, PDAC-3, Blood-1, Blood-2, Blood-3) to its corresponding start ratio (Enrich-2), and subsequently ordered from highest to lowest fold change based on PDAC-3 (for PDAC-1 and PDAC-2) or Blood-3 (for Blood-1 and Blood-2). Finally, the results were filtered to remove all unique peptides with a fold increase of either ≥1, ≥10, or ≥50 in Blood-3 from PDAC-3. To gain more insight into the amino acid distribution during the experiment, the ratio of every amino acid for every position in each unique peptide in all samples was determined, and fold change in amino acid was determined by normalization of the ratio in a given sample to the start ratio (Enrich- 2). Finally, samples were compared by calculating differences in fold change of Enrich-2-normalized amino acid ratios.
FRET-peptide cleavage assay
FRET-peptides containing substrate sequences corresponding to the top 15 hits and the CAPN2-responsive substrate, SGAGLPLFAARPGANS, were synthesized by modifying the peptide with Dabcyl at the N-terminus and a FAM-group at the C-terminus (LifeTein, Somerset, NJ, USA). Next, 10 μL of 10 μM peptide was incubated with 90 μL of PBS (negative control), PANC-1, MIA PaCa-2, BxPC-3, Capan-1, Capan-2, human blood lysate (1 × 106 cells/mL) or murine lung, liver, kidney or colon lysate. Fluorescence was measured every 15 min at Ex/Em 485/528 nm wavelengths using a Biotek Synergy HT plate reader (Biotek Instruments, Winooski, VT, USA).
Statistical analysis
All statistical tests, sample sizes, and error bar definitions are given in the respective figure legend. Statistical tests were conducted in GraphPad Prism (version 9.1.0, GraphPad Software Inc.), and graphs were made in GraphPad Prism (version 9.1.0, GraphPad Software Inc.).







留言 (0)