
記住我
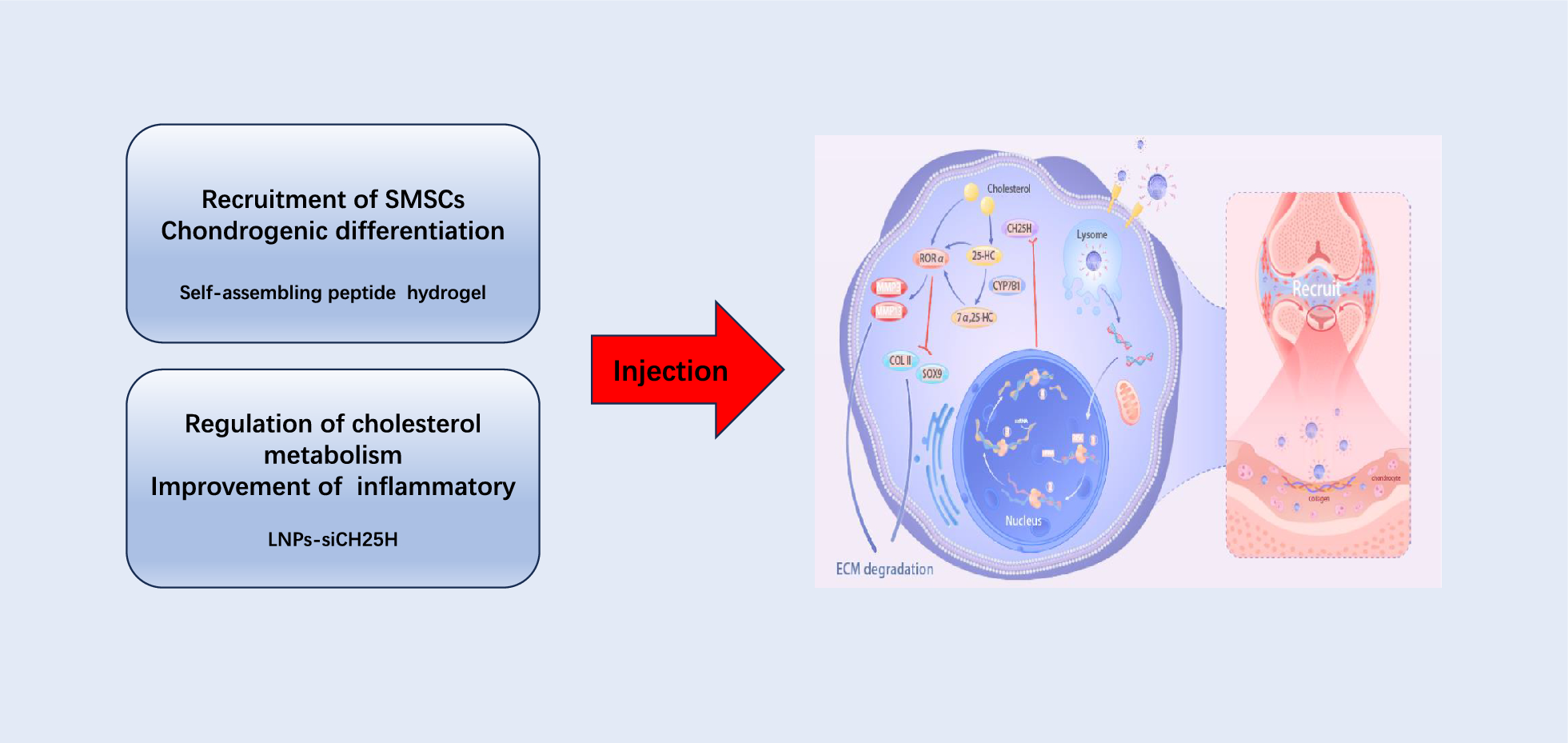
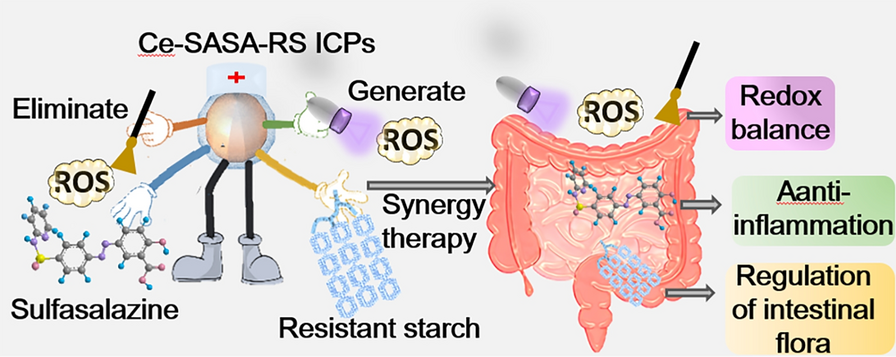
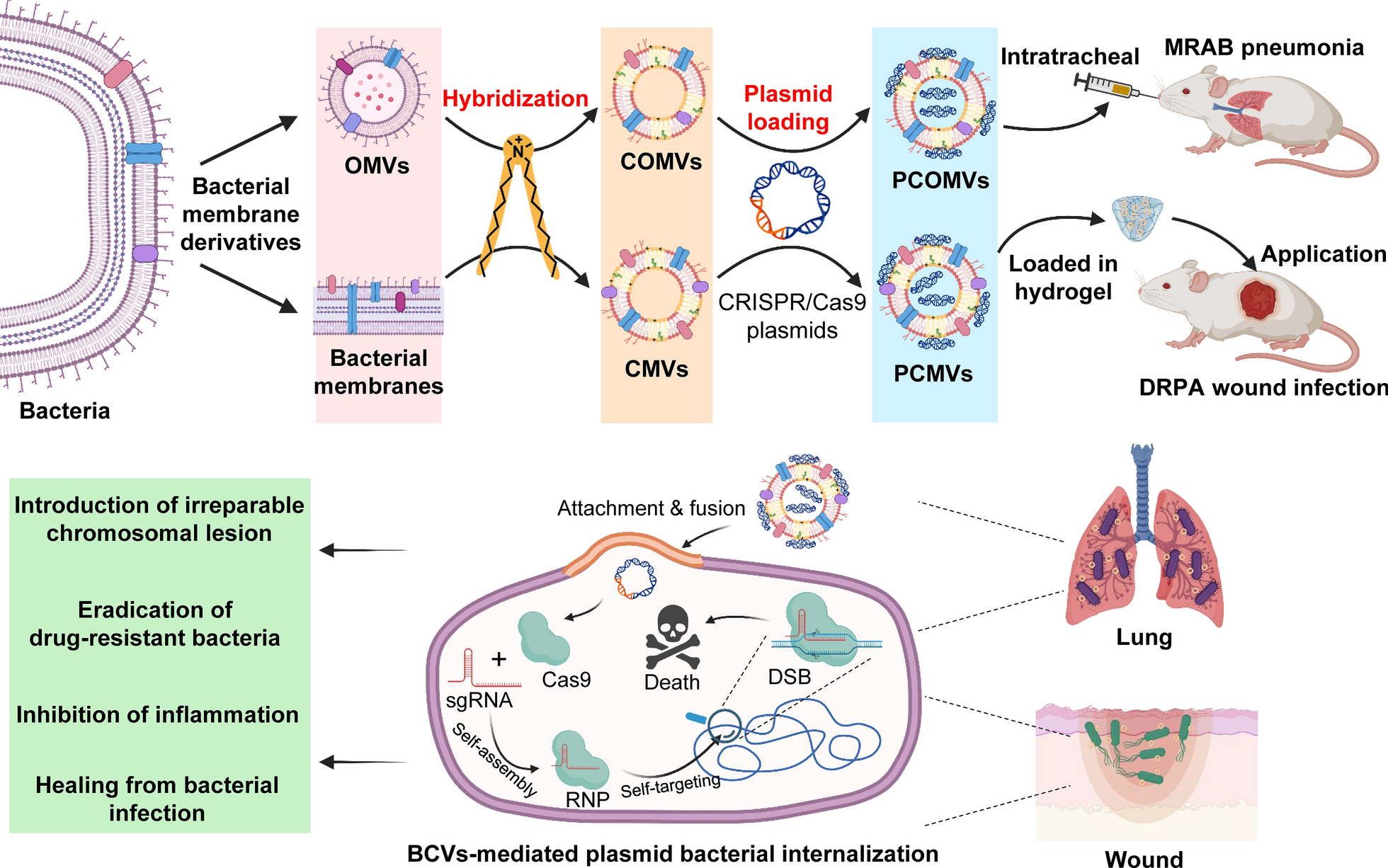






As the most commonly used biological evaluation method, both the bone regeneration effects in vivo and cellular experiments in vitro play an important role in the functional study of biomaterials. However, little is known of the fate and bioactivity of cell response to the implant. It is imperative to decipher the distinct fate of stem cells after scaffold therapy during the regeneration of defective bone. This study was aimed to develop a strategy to identify endogenous bone regeneration mechanism triggered by gastrodin-PU/n-HA. The scaffolds were prepared as osteoinductive implants to guide the regeneration of osteoporotic bone defects. The scaffolds were then implanted into condyles of femur in an osteoporotic rat model. The changes in serum and tissue inflammatory markers were first monitored during implantation. Furthermore, sequential cell extraction was carried out to explore the cell recruitment and reveal the time course of stem cell fate, focusing on the balance between stem cell population and osteo-differentiation via ACD. New bone regeneration within and adjacent to the scaffolds was finally assessed by micro-computed tomography (micro-CT) scanning and histological staining. Subsequently, histological, immunohistochemical (IHC) staining were conducted to explore the potential osteogenic mechanism of the scaffolds.
Preparation and characterization of scaffolds Fabrication of scaffoldsScaffolds were successfully synthesized through in situ foaming method according to our previously reported method [18]. In brief, PCL-2000 (30.00 g) and n-HA (11.40 g) were mixed in a 250 mL three-necked flask under nitrogen continuously and heated at 70 ℃, stirred with IPDI (9.17 g) for 2 h to obtain the prepolymer. Then 3.70 g of Lys-OEt·2HCl was used to extend the prepolymer for 2 h. After that, gastrodin of different contents (set as 0, 2, and 5 wt%) was added, followed by 1 mL NaHCO3 solution. The resultant mixture was cured at 90 ℃ for subsequent use.
CharacterizationFor the degradation and gastrodin release study, the scaffolds (~ 0.20 g) were immersed in 0.1 M and 0.5 M NaOH solution at 37 ℃ water bath, respectively. At 3/7, 1, 2, 3, 4, 5, 6, 7, 8 weeks, the residual weight and released gastrodin of each specimen were recorded. Micro-CT (Hiscan, China) and scanning electron microscopy (SEM) (Zeiss, Germany) were carried out to observe the microstructure of the scaffolds, and the porosity was quantified by dragonfly software (ORS, Canada). Mechanical properties were measured on a universal testing system (Instron, USA).
Surgical proceduresThe scaffolds were cut into cylinder (Ø 3 × 3 mm) and sterilized by γ-ray irradiation with 15 kGy prior to use. The animal experiments were approved by the Institutional Animal Care and Use Committee of Kunming Medical University (China). Female Sprague-Dawley (SD) rats weighing 200 to 300 g were anesthetized with 3% sodium pentobarbital (30 mg/kg) and subjected to a standard bilateral ovariectomy (OVX) as described [28]. Three months after OVX, the osteoporotic rats were randomized into three groups: control, PU/n-HA, and 5% gastrodin-PU/n-HA. After exposure of lateral femoral condyle, a drill-hole defect with a diameter of 3 mm and a depth of 3 mm was created under abundant irrigation. The bone fragments were washed out from the cavity before the implantation of scaffolds, followed by repositioning and suturing of the tissue layers. The control group was operated without implantation of scaffolds. At postoperative days 3, 7, 14, and 28, rats were anesthetized for the peripheral blood harvesting. After sacrificing by cervical dislocation, bone marrow, and femur were also harvested for subsequent analysis.
Stem cells preparationBone marrow mesenchymal stem cells (BMSCs) were isolated from femurs according to our previously reported procedure [18] with minor revisions. At postoperative days 7 and 14, the femurs were soaked in 75% ethanol for 10 min and then washed with phosphate buffer saline (PBS, Solarbio, China). After both ends of the femur were removed, a disposable aseptic syringe drawing a complete medium consisting of α-minimum essential medium (α-MEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin/streptomycin (Gibco, USA) was used to repeatedly wash the bone marrow cavity. About 4 mL of BMSCs suspension was obtained from two femurs of a rat, which was split into three portions: centrifuge tube (300 µL), T25 cell culture flask (300 µL), and culture plates (100 µL in 24-well plate and 200 µL in 6-well plate). Next, the BMSCs transferred to culture flasks and culture plates were further incubated at 37 °C, 5% CO2, and 95% humidity. The complete medium was used for follow-up experiments and changed every 3 days unless otherwise stated.
For long-bone-derived skeletal stem cells (SSCs), femurs were dissected. After flushing out the bone marrow cells, the femurs with implants were then chopped into small pieces and digested by collagenase II (Sigma, U.S., 1 mg/mL) at 37℃ for 1 h. The digested cells were cultured in complete medium and allowed for functional assessment (See Scheme 2).
Scheme 2
Schematic illustration of the experimental outline. SD rats were subjected to a standard bilateral ovariectomy (OVX) and scaffolds were implanted into femoral condyle defect after 3 months. The stem cells were isolated and collected at 7 and 14 days, and further incubated for 1, 3, 5, 7, and 14 days for analysis
Endogenous stem cell recruitmentFlow cytometry (FC)BMSCs in each tube were filtered with a 70 μm strainer to yield single cell suspensions. The cells were then incubated with red blood cell lysis buffer on ice, after which they were centrifugated and resuspended in FC sorting buffer (PBS with 10% FBS). Cells were stained with armenian hamster anti mouse/rat monoclonal antibodies CD29-APC (No.: 102215), mouse anti-rat monoclonal antibodies CD45-PE/CY7 (No.: 202213), and CD90-PE (No.: 205903) (Biolegend, USA) (1:200) according to the manufacturer’s protocol. FC was performed using CyFlow® Space (Partec, Germany), and data analysis was conducted with FlowJo 10.5.3 software.
EdU incorporation assaysBMSCs were seeded in 24-well plates (100 µL/well) and maintained in culture. At 3 and 7 days, exponentially growing cells were stained with a Click-iT 5-ethynyl-2’-deoxyuridine (EdU) Alexa Fluor 594 Imaging Kit and nuclei were stained with Hoechest (Beyotime, China). Images were captured using a fluorescence microscope (Olympus, Japan). The numbers of EdU-positive cells were further counted by ImageJ software. The percentage of proliferating cells was calculated for each sample relative to the total number of Hoechest-positive cells.
Colony-forming unit fibroblast formation assay (CFU-F assay)Self-renewal of BMSCs was assessed by a CFU-F assay. After being cultured in a 24-well plate (100 µL/well) for 7 days, cells were fixed and stained with crystal violet solution (Beyotime, China).
Transwell-migration assayA transwell chamber (24-well plate, 8 μm pore size, Corning Incorporated, USA) was used to evaluate the cell migration of retrieved scaffolds. The retrieved scaffolds were achieved from implants within osteoporotic bone defects or subcutaneous tissue after 7 and 14 days. Healthy 3-week-old rats served as donors of primary BMSCs, which were subcultured to the third passage (P3 BMSCs) according to the established procedure. Subsequently, 0.1 mL P3 BMSCs suspension (5 × 104 cells/mL) was added to the upper chamber. On the other hand, the retrieved scaffolds were placed in the lower chamber with 0.6 mL complete media. After 3 and 7-day incubation, the cells were fixed with 4% paraformaldehyde and stained with crystal violet. The non-invaded cells on the upside of the transwell were removed with a cotton swab, and the migrated cells to the underside were examined in three random fields under a light microscope. Meanwhile, the medium was collected and used for evaluation of secretion of SDF-1α and TGF-β cytokines with enzyme-linked immunosorbent assay (ELISA) kits (mlbio, China) following the manufacturer’s guidance.
Functional assessment of stem cellsMitochondrial activity of BMSCsBMSCs were cultured with complete medium in 24-well plates for 7 and 14 days. At each time point, samples were stained with MitoTracker® Red CMXRos (Solarbio, China, No.: M9940) for active mitochondria. After fixation with 4% paraformaldehyde solution, the samples were stained with phalloidin (Solarbio, China, No.: CA1640) for actin and DAPI (Abcam, USA, No: ab104139) for nuclei. The mitochondrial membrane potential was detected using a probe 5,5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) assay kit (Solarbio, China, No.: J8030) according to the manufacturer’s instructions.
For immunofluorescence (IF) staining, the wells were washed and fixed with 4% paraformaldehyde for 15 min at room temperature. The samples were then permeabilized in 0.1 % Triton X-100 for 20 min. Next, 5 % goat serum in PBS was added to the wells for 2 hours, followed by exposure to primary antibody cocktail, which was incubated overnight at 4 °C. The primary antibody involved mitochondrially encoded cytochrome C oxidase 2 (Mtco2), translocase of the outer mitochondrial membrane (TOM20), ATP synthase subunit alpha (ATP5A1), peroxisome proliferator-activated receptor-gamma coactivated 1α (PGC1α). Mtco2 (1:200), TOM20 (1:200), cocktail ATP5A1 (1:1000) and PGC1α (1:200) in 5% goat serum in PBS were used. The secondary antibodies were as follows: CoraLite488-conjugated goat anti-rabbit IgG (1:500, SA00013-2), CoraLite594-conjugated goat anti-rabbit IgG (1:200, SA00013-4), CoraLite594-conjugated goat anti-rabbit IgG (1:200, SA00013-4) and CoraLite488-conjugated goat anti-mouse IgG (1:500, SA00013-1). Mean optical density of the proteins was measured by Image J software. Each group comprised more than three wells, and each well was confirmed with cells collected from at least three separate animals. The related genes and proteins were further assessed as stated above.
Immunofluorescence staining of cellsBMSCs were seeded on coverslips in 24-well plates (100 µL/well) and cultured for 1, 3, 5, and 7 days. IF staining was carried out as indicated in the section IF staining of mitochondria. The following primary antibody cocktails in 5% goat serum in PBS were used: octamer-binding transcription factor 4 (OCT4) (1:200) and osterix (OSX) (1:200), β-catenin (1:2,000) and atypical protein kinase C (aPKC) (1:200). After rinsing with 0.05% PBST, the wells were incubated with secondary antibody cocktails for 2 h at room temperature. The secondary antibodies were as follows: CoraLite488-conjugated goat anti-rabbit IgG (1:500) and CoraLite594-conjugated goat anti-mouse IgG (1:200), CoraLite488-conjugated goat anti-mouse IgG (1:500) and CoraLite594-conjugated goat anti-rabbit IgG (1:200), of which the former corresponded to OCT4/OSX and the latter to β-catenin/aPKC cocktails. Finally, DAPI staining was performed for 2 h at room temperature. In parallel, the SSCs were also processed for the aforementioned IF staining.
Imaging and analysis of ACD processAll the imaging stacks were captured and processed using a confocal laser scanning microscope (CLSM) (Zeiss LSM 880, German) and Zen blue edition 3.3 software attached to the microscope. Briefly, z-stacks with 20 z-planes were acquired consecutively at a 1-µm z-step for each volume. Each z-stack was 3D-reconstructed and presented maximum intensity projections onto frontal X/Y, transverse (X/Z), and sagittal (Y/Z) planes. To calculate the proportion of the asymmetric stem cell division, 20 cell pairs were blindly chosen from each sample. The mean fluorescence intensity and distribution of the protein markers in paired daughter cells were measured, followed by normalization between the two newly formed daughter cells.
ACD-related gene expressionAfter the cells were subsequently cultured for 3 and 7 days, the ACD-related genes such as OCT4, OSX, β-catenin and aPKC were evaluated by reverse transcription quantitative polymerase chain reaction (RT-qPCR). The primer sequences are described in Table S1. Each sample was conducted in triplicates and repeated for three independent assays to achieve comparable results.
Endogenous new bone formationInflammatory/immune response analysisAt postoperative days 3, 7, 14, 28, the peripheral blood was collected in anticoagulant tubes for analysis using hematology analyzer (Sinnowa, China). The serum was derived from peripheral blood by centrifugation at 4000 rpm for 15 min. The content of inflammatory factors (Tumor Necrosis Factor-α (TNF-α) and interleukin-1β (IL-1β)) and reactive oxygen species (ROS) level in serum were detected by ELISA assay kit (Nanjing Jiancheng, China) according to the manufacturer’s instructions. Tissue samples from the surrounding areas were collected and stored at -80 ℃ for the follow-up experiments. Total mRNA was extracted from bone tissue for analysis of macrophage phenotype biomarker genes, inducible nitric oxide synthase (iNOS) and arginase-1 (Arg-1).
Histological staining of tissue sectionsThe fixed samples were decalcified in 10% EDTA solution for 1 month, dehydrated through gradient ethanol, and then embedded in paraffin. Serial 5-µm thick sections were cut on a microtome (Leica Biosystems, Germany). The tissue sections were stained with Hematoxylin and eosin (H&E) (Solarbio, China), Masson’s trichrome (Solarbio, China), and Movat’s pentachrome (Solarbio, China) according to the supplied method by the manufacturer. Representative section images were captured with a light microscope (Olympus, Japan).
Immunohistochemistry staining of tissue sectionsTissue sections were incubated with a 3% H2O2 solution in methanol for endogenous enzyme quenching and treated with 0.1% trypsin-EDTA (Gibco, 25200056) to retrieve the antigen. After blocking with 5% goat serum for 1 h at 37 °C, the specimens were incubated overnight at 4 °C with primary antibodies against CD146 (1:200) for stem cell surface markers, BMP-2 (1:200) for osteogenesis markers, Arg-1 (1:200) for an M2 marker, and iNOS (1:200) for an M1 marker, as listed in Table S2. The sections were washed with TBST before incubation with the secondary antibodies for 2 h. Sections were further incubated with secondary antibodies (Enhanced enzyme-labeled goat anti-mouse IgG polymer and Enhanced enzyme-labeled goat anti-rabbit IgG polymer), and then with DAB reagents (Diaminobenzidine tetrahydrochloride staining, Boster Biological Technology Co. Ltd., China), followed by counterstaining with hematoxylin. The sections were mounted prior to imaging. Brown staining localization within the defects indicated positive expression of the biomarkers under an optical microscope. The positive cell ratio of the proteins was calculated by software Image J and GraphPad Prism v8.0. Each type of staining was confirmed in tissues from more than three separate rats.
Micro-computed tomography (micro-CT) analysisFour weeks after implantation, femurs were collected and fixed in 4% paraformaldehyde for 3 days. The fixed samples were scanned using micro-CT (Pingseng scientific, China) at a resolution of 7.5 μm with a voltage of 90 kV and a current of 60 µA. During the reconstruction, a threshold between 1000 and 3000 was applied to discriminate the bone tissue, and a threshold between 600 and 1000 was applied to discriminate the scaffold. Three-dimensional (3D) and two-dimensional (2D) analysis were performed using Avatar3 software attached to the micro-CT. 3D reconstructed structures of the distal femur were acquired from the segmented dataset for visual inspection. For the quantitative evaluation of osteogenesis within defects, a consistent cylinder at a diameter of 2.5 mm and a height of 2.5 mm was chosen as the region of interest (ROI) to represent remnant scaffold and regenerated bone. To study the effect of the scaffolds on adjacent bone loss, another rectangular ROI (0.5 × 0.5 × 2 mm3) adjacent to the defect, which was 1.5 mm away from the epiphyseal growth plate, was also selected and analyzed. The structural parameters of new bone and trabecular bone, including bone mineral density (BMD), trabecular bone volume fraction (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular separation (Tb.sp), were calculated through the micro-CT scans.
Statistical analysisData were reported as means ± standard deviation (SD). One-way analysis of variance (ANOVA) was applied for statistical comparisons among more than two groups, and a t-test was used for statistical comparisons between two groups. All statistical analyses were performed using GraphPad Prism v8.0.2. Statistical significance was accepted at P < 0.05.
留言 (0)