Mutant selective inhibitorsTargeting KRAS G12C
Although the KRAS G12C mutation is found in only 1.6% of PDAC patients, it is the only KRAS mutation for which selective inhibitors have reached clinical application. Historically, the KRAS protein was considered “undruggable” until 2013, when Shokat and colleagues identified a novel cysteine-containing switch II pocket in KRAS G12C [7, 8]. The CodeBreaK 100 trial, a multicenter phase I/II study, evaluated sotorasib, a selective covalent inhibitor of KRAS G12C. Among 38 patients with KRAS G12C-mutated metastatic PDAC, the confirmed objective response rate (ORR) was 21%, with a median progression-free survival (mPFS) of 4.0 months and a median overall survival (mOS) of 6.9 months [9]. The KRYSTAL-1 study investigating adagrasib showed an ORR of 33.3%, a disease control rate (DCR) of 81%, an mPFS of 5.4 months, and an mOS of 8 months for 21 PDAC patients [10]. Several other KRAS G12C inhibitors, such as divarasib, olomorasib, and glecirasib, are currently in (pre)clinical evaluation, demonstrating ORRs of up to 40% in pancreatic cancer cohorts (Table 1; [11,12,13]). Further studies are needed to validate the clinical benefit of these targeted therapies in the PDAC population and to assess the efficacy of combining them with other treatments.
Table 1 Outcome of reported efficacy and safety data of various KRAS G12C inhibitors in PDACTargeting other KRAS mutants
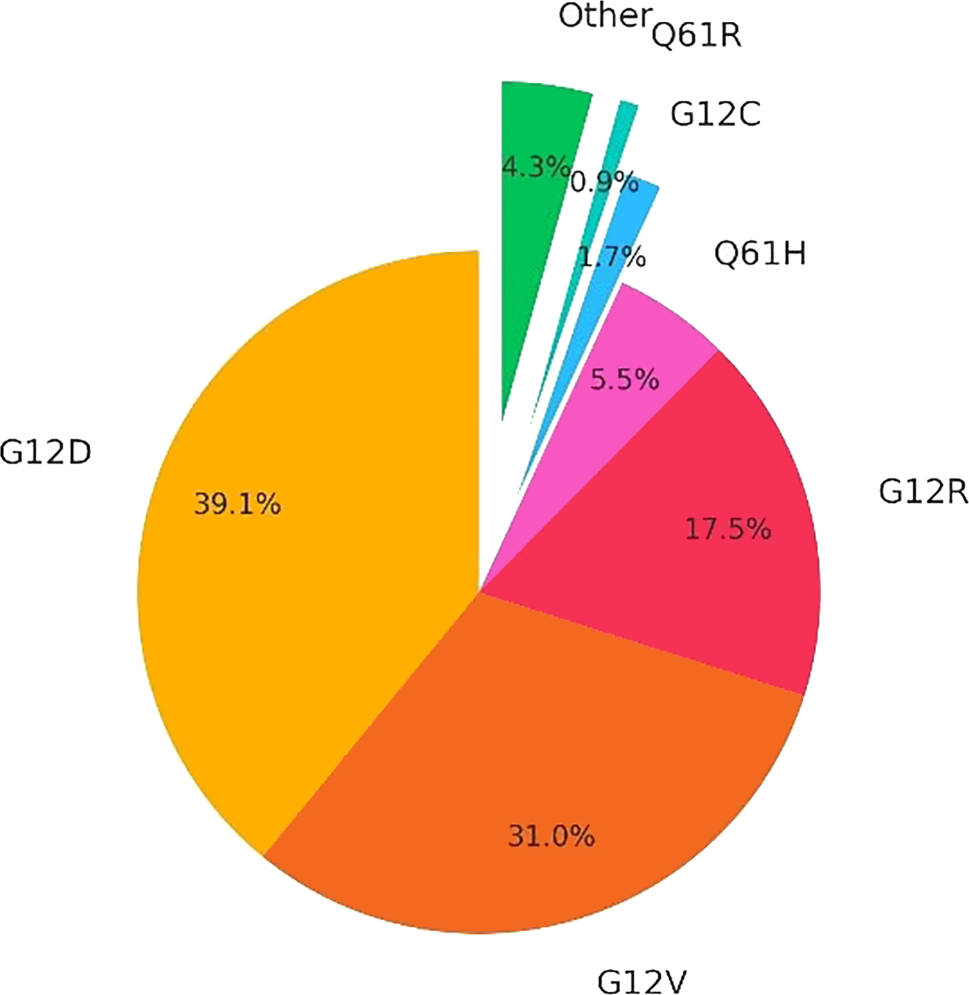
The most prevalent KRAS mutation in PDAC is KRAS G12D, making it a critical target for therapeutic intervention. However, the switch II pocket of the KRAS G12D protein lacks a reactive cysteine, making it more difficult to target with covalent inhibitors. In 2021 Wang et al. developed MRTX1133, a non-covalent KRAS G12D inhibitor based on the structure of adagrasib. Preclinical data demonstrate that MRTX1133 can reverse early PDAC growth and positively influence the tumor microenvironment (TME). Additionally, RMC-9805, a covalent tri-complex RAS(ON) G12D-selective inhibitor, has shown promise in preclinical PDAC models and was presented at AACR 2023 [17]. Moreover HRS-4642, another KRAS G12D inhibitor, has entered a first-in-human phase 1 trial, with early findings presented at ESMO 2023 [18].
Development of rapid resistance in mutant-specific targeting
The response duration to KRAS G12C inhibition in PDAC is typically modest, with resistance developing early. Acquired resistance to KRAS inhibition occurs primarily through three mechanisms [19, 20]:
1.
Acquisition of additional KRAS mutations: For example, in KRAS codons 12, 13, or 61, leading to enhanced RAS-MAPK pathway activation or disruption of binding at the switch II pocket.
2.
Bypassing KRAS inhibition through alterations in other RAS isoforms: Secondary mutations in other RAS family members (NRAS, HRAS) can bypass KRAS inhibition, sustaining oncogenic signaling.
3.
Alterations in downstream effector kinases: Mutations in downstream signaling components, such as BRAF, MEK1, NF1, PTEN, or PI3K, can also contribute to resistance by reactivating the signaling pathways despite KRAS inhibition.
In addition to acquired resistance mechanisms, different KRAS G12 mutations exhibit innate resistance to inhibitors based on the cycling velocity between ON and OFF states. Mutations with a very slow or negligible rate of GTP hydrolysis, such as KRAS G12V or G12D, may be “locked” in the ON state, potentially rendering them refractory to OFF-state KRAS inhibitors. ON-state inhibitors block effector engagement more rapidly, while OFF-state inhibitors may require longer to achieve complete target engagement and suppression, raising concerns about their efficacy in these cases [21].
Strategies to overcome mechanisms of acquired resistanceTargeting KRAS upstream pathways
Indirect pan-KRAS inhibition can be achieved by targeting upstream signaling proteins involved in KRAS activation, specifically SHP2 and SOS1 (Son of Sevenless 1). Several SHP2 inhibitors are in development, with some advancing to clinical trials. These include AB-331, presented at ASCO 2024, as well as RMC-4630, TNO155, and JAB-3068, which are being evaluated in phase I/II clinical trials (NCT03634982, NCT04330664, and NCT05288205; [22]). Additionally, BI1701963, a SOS1 inhibitor, is under investigation in a phase I trial to determine dosing as monotherapy and in combination with trametinib for KRAS-mutated solid tumors (NCT04111458).
Pan-KRAS inhibitors
Isoform-selective targeting of RAS may offer a promising approach by inhibiting all KRAS mutants, potentially addressing secondary activating KRAS mutations as well. BI-2865, the first non-covalent pan-KRAS inhibitor, has demonstrated preclinical efficacy against tumors harboring common KRAS alterations. BI-2865 specifically binds to the GDP-bound OFF state of both KRAS WT and its mutant variants, while sparing NRAS WT and HRAS WT isoforms, which may be important for maintaining tolerability in patients [23]. However, a critical challenge for the clinical application of pan-KRAS-selective inhibitors is the limited understanding of the effects of inhibiting KRAS WT in humans, raising concerns about potential adverse consequences.
Pan-RAS inhibitors
The primary challenge with pan-RAS inhibition is the potential toxicity from off-target effects on RAS WT in normal tissues, which could involve all three RAS isoforms (KRAS, HRAS, and NRAS). Preliminary data presented at ESMO 2023 from a phase I trial of the first-in-class pan/multi-KRAS inhibitor RMC-6236, a RAS-selective tri-complex, showed promising results in patients with previously treated metastatic PDAC (n = 46), with an ORR of 20% and a DCR of 87% [24]. Additionally, RMC-7977 has demonstrated efficacy across various preclinical pancreatic cancer models [25]. Both RMC-6236 and RMC-7977 function as tri-complex inhibitors that require cyclophilin A to target the GTP-bound active (ON) state of wild-type and mutant isoforms of KRAS, NRAS, and HRAS. Safety concerns might be mitigated by the enrichment of cyclophilin A in tumor cells or the ability of RMC-7977 to specifically target the GTP-bound ON state of RAS.
Other (K)RAS-directed approachesCellular therapies
Encouraging data are emerging for TCR therapy in highly select patients with PDAC. A case report of autologous T‑cell therapy expressing HLA-C * 08:02–restricted TCRs targeting KRAS G12D indicated efficacy in two patients with chemotherapy-refractory PDAC, with one patient maintaining a durable PR lasting more than 6 months [26]. There are several ongoing phase I/II clinical trials evaluating TCR therapy directed against KRAS G12V (NCT04146298, NCT03190941) and KRAS G12D (NCT03745326) in PDAC.
KRAS vaccines
Vaccination offers potential to expand endogenous KRAS G12-directed T cells across diverse HLA backgrounds, with simplified manufacturing and off-the-shelf availability. So far, conventional peptide vaccines suffered from poor immunogenicity. By contrast, modifying antigens—KRAS G12D and KRAS G12R peptides—and adjuvants—Toll-like receptor 9 agonists—with diacyl lipids that associate with fatty-acid-binding pockets on endogenous albumin after injection (creating Amph vaccines) results in efficient delivery into antigen-presenting cells. The first results in the adjuvant setting (ELI-002 2P vaccine; AMPLIFY-201 trial) led by Prof. O’Reilly from the Memorial Sloane Kettering Cancer Center showed a biomarker reduction in 16 out of 20 and MRD biomarker or circulating DNA (ctDNA) clearance in 3 out of 20 cases [27]. Vaccines utilizing mRNA technology for KRAS-mutated PDAC include mRNA-5671, which encodes KRAS G12D-, G12V-, G13D-, and G12C-specific peptides, are currently under investigation in combination with pembrolizumab in a phase I trial in KRAS-mutant advanced cancer (NCT03948763).





留言 (0)