Cell cultures and treatment with sorafenib
The following BC cell lines were used in this study: HCC1937 (ATCC CRL-2336), MDA-MB-453 (ATCC HTB-131), MCF-7 (ATCC HTB-22), and MDA-MB-231 (ATCC HTB-26). Cells were maintained in RPMI-1640 (Thermo Fisher Scientific); all culture media were supplemented with 20% (HCC1937 cells) or 10% (MDA-MB-453, MCF-7, and MDA-MB-231 cells) of Fetal Bovine Serum (Thermo Fisher Scientific) and 10,000 U/ml penicillin/streptomycin (ThermoFisher Scientific); cells were grown at 37 ℃ with 5% CO2. Sorafenib was synthesized and provided by Bayer Corporation (West Haven, CT, USA). This compound was dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich) and diluted with RPMI-1640 to achieve the required concentration. 0.1% DMSO was added to the cell cultures as a solvent-only negative control for in vitro studies.
EVs isolation
All cell lines were seeded in number of 300,000/ 10 cm Ø dish, in order to isolate EVs via three methods. The first method employed the use of Total Exosome Isolation Reagent (from cell culture media) (Thermo Fisher Scientific) as follows: cells were seeded in a 10 cm Ø dish until they reached 90% confluence. The cells monolayer was washed 3 times with 1x PBS and 15µM sorafenib or 0.1% DMSO, was added as treatment for 24 h, in the absence of FBS; cell culture media represented by RPMI-1640 (- FBS) was harvested, centrifuged at 2,000 x g for 30 min to remove the remaining cells and debris, and the supernatant was transferred into a new tube without disturbing the pellet; afterward, the appropriate volume of Total Exosome Isolation reagent was added (500 µL of reagent per 1 mL of cell media), vortexed until obtaining a homogenous solution and incubated overnight at 4 ℃; following incubation, tubes were centrifuged at 10,000 x g for 1 h at 4 ℃, the supernatant was discarded without disturbing the pellet (not visible), where EVs were found, and resuspended in 1X PBS (for 10 mL of cell media 200 µL of 1X PBS were required). For the second used method, EVs were purified by ultracentrifugation as indicated by the International Society of Extracellular Vesicles (ISEV) [28]. Briefly, the conditioned medium was centrifuged at 300 g for 10 min at 4 °C to pellet the cells. The supernatant was collected and centrifuged at 16,500 x g for 20 min at 4 °C to eliminate apoptotic bodies and cell debris. The supernatant was then filtered through 0.22 μm filters and ultracentrifuged at 110,000 x g for 70 min at 4 °C. Pellets were resuspended in 1x PBS and ultracentrifuged at 110,000 x g for 70 min at 4 °C. Finally, the pellet containing EVs was resuspended in 1x PBS [29]. As a third method, EVs were purified by immunoprecipitation using the Exosomes Isolation Kit Pan (Miltenyi Biotec, Germany) following the manufacturer’s instructions. EVs obtained with the first and second methods were quantified by BCA Protein Assay Kit (Thermo Fisher Scientific). In contrast, immunoprecipitated EVs were not quantified because magnetic beads interfere with quantification methods.
Western blotting
EVs were lysed in Laemmli Buffer (100 mM Tris–HCl pH 6.8, 4% SDS, 20% glycerol, and 0.2% blue bromophenol) and then quantified using the BCA assay (Thermo Fisher Scientific). Thus, the proteins of EVs were separated by SDS-PAGE, and precisely 2 µg of EV lysate obtained with the precipitation method was loaded. For immunoprecipitated EV, the number of cells was used as the normalization parameter. Subsequently, electroblotting onto polyvinylidenefluoride (PVDF) Immobilon-P membranes was performed (Millipore, Billerica, MA, USA), and the membrane was blocked in 5% milk in PBS for 30 min at room temperature [32]. Primary antibodies were incubated overnight and anti-mouse and anti-rabbit peroxidase-conjugated secondary antibodies were incubated for 1 h at room temperature. The presence of the cytosolic protein, Tumor Susceptibility gene 101 (TSG101), and three tetraspanins CD63, CD9, and CD81 (positive controls), and the absence of the non-EV component Ribosomal Protein S6 (RPS6) (negative control) were verified.
Transmission electron microscopy (TEM)
TEM analysis of isolated EVs was performed with a JEOL JEM-1011 transmission electron microscope at 100 kV operating voltage, equipped with a 7.1 megapixel CCD camera (Orius SC1000, Gatan, Pleasanton, CA). TEM image analysis was achieved with Gatan Digital Micrograph™ (DM) software. For sample preparation, a 5 µL drop of a concentrated vesicle suspension was dropped on a Formvar-coated copper grid (placed on parafilm); after 20 min the grid was upside down infiltrated onto a 50 µL drop of cold carboxymethyl dextran solution (1 mg/mL in UP water) for 5 min. The excess liquid was slowly adsorbed tangentially by using Grade 1 Whatman paper. The resulting ultrathin polysaccharide layer prevents the vesicle collapse on the dried grids. The above-described protocol was slightly modified from the one used by Kreger B.T. et al. [30]. The grids were finally stained by UranyLess EM Stain (Electron Microscopy Sciences), by following the standard protocol provided by the manufacturer.
Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was performed using the Nanosight NS300 instrument (Malvern). A standard operating procedure was applied to acquire three independent videos of 60 s of each sample under a syringe pump speed 30. Samples were systematically diluted in 1x PBS to reach the recommended number of particles per frame and were recorded at Camera level 11. Particle analysis was performed with a detection threshold 5.0, always using NanoSight NS300 software NTA 3.4 Build 3.4.003 (Malvern).
RNA isolation and reverse transcription (RT)
Total RNA was isolated from 200 µL of EVs resuspended in 1x PBS (Thermo Fisher Scientific) using miRNeasy Mini Kit (Qiagen), according to the manufacturer’s instructions. TRIzol RNA Isolation Reagent (Thermo Fisher Scientific) was used for the total RNA isolated from cell cultures, according to the manufacturer’s instructions.
For lncRNA analysis, cDNA was synthesized from 5 µL of total RNA from EVs and 1 µg of intracellular RNA, in a 20 µL reaction volume, using M-MLV Reverse Transcriptase (Sigma-Aldrich) according to the manufacturer’s instruction. The RT reaction was performed at 70 ℃ for 10 min, followed by incubation at room temperature for 10 min, a second incubation at 37 ℃ for 50 min, followed by inactivation at 94 ℃ for 10 min in a T100 Thermal Cycler (Bio-Rad Laboratories).
For miRNAs analysis, cDNA was synthesized from 5 µl of RNA from EVs and 50 ng of cellular RNA, in a 15 µL reaction volume, using the TaqMan microRNA Reverse Transcription Kit components (Thermo Fisher Scientific) and the stem-loop primer for miR-23b-3p (Thermo Fisher Scientific; Assay ID 000400) and miR-126-3p (Thermo Fisher Scientific; Assay ID 002228). RT reaction was performed at 16 ℃ for 30 min, 42 ℃ for another 30 min, followed by inactivation at 85 ℃ for 5 min in a T100 Thermal Cycler (Bio-Rad Laboratories).
Droplet digital PCR workflow
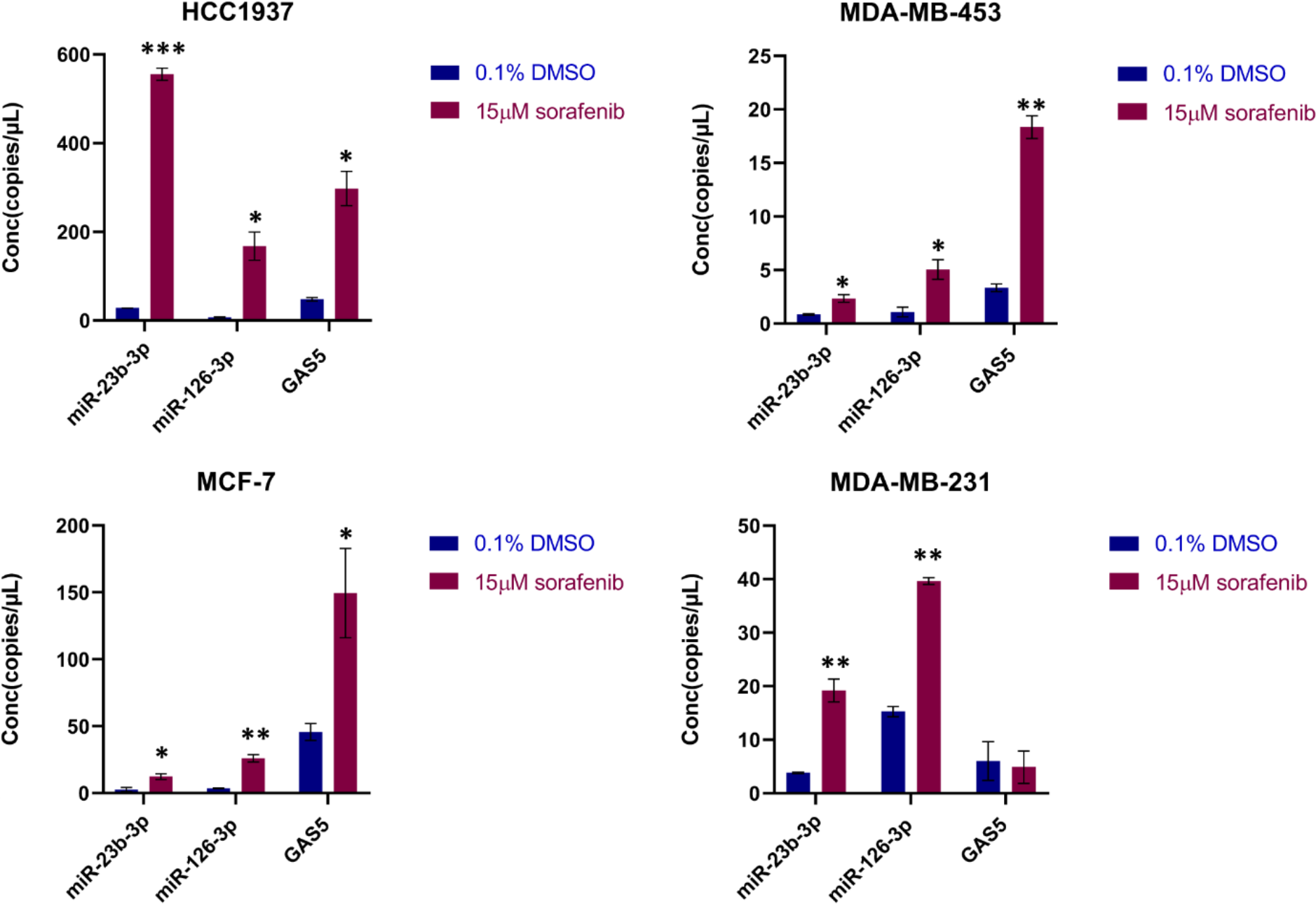
The synthesized cDNA was used as a template for the ddPCR experiments using the QX200 Droplet Digital PCR (ddPCR) System (Bio-Rad Laboratories), and ddPCR was performed according to the ddPCR Supermix for Probes (Bio-Rad Laboratories) protocol [31]. Briefly, 1.33 µl of the cDNA obtained using TaqMan microRNA Reverse Transcription Kit, and 3.96 µl of the cDNA obtained using M-MLV Reverse Transcriptase, were prepared for amplification in a 20 µL reaction volume containing 2x ddPCR Supermix for Probes (Bio-Rad Laboratories), 20x TaqMan assay (Thermo Fisher Scientific) specific for miR-23b-3p, miR-126-3p, and PrimeTime qPCR Assay specific for the lncRNA GAS5 (Integrated DNA Technologies) and water. Each ddPCR assay mixture (20 µL) was loaded into a disposable droplet generator cartridge (Bio-Rad). Then, 70 µL of droplet generation oil for probes (Bio-Rad) were loaded into the wells dedicated to oil. The cartridge was then placed inside the QX200 droplet generator (Bio-Rad). When droplet generation was completed, the droplets were transferred to a 96-well PCR plate using a multichannel pipette. The plate was heat-sealed with foil and placed in a T100 Thermal Cycler (Bio-Rad Laboratories). A negative control (NC) and a positive control (PC) were included. Concentration data for miR-23b-3p, miR-126-3p and GAS5 levels were obtained using QuantaSoft Software (Bio-Rad Laboratories) as copies/µL.
EVs labeling and uptake by target cells
MDA-MB-453 and MCF-7 BC cells were incubated with CellTracker™ CM-DiI Dye (Thermo Fisher Scientific) diluted 1:1,500 in RPMI (-FBS) for 24 h at 37 ℃ with 5% CO2 and labeled EVs were obtained. Coverslips were placed into 3 cm diameter plates and 3 × 105 cells were seeded and incubated at 37 ℃ with 5% CO2. When indicated, labeled EVs were added to the cells in a final volume of 3 mL of RPMI (-FBS) and incubated for 24 h at 37 °C. Samples were washed with 1x PBS, fixed with methanol 100% and the nuclei stained with DAPI (1:3,000, Merck, Inc., Darmstadt, Germany) for 15 min. After washing with 1x PBS, the coverslips were mounted with Vectashield mounting medium (Vector Labs, Newark, CA, USA) and visualized using a Leitz fluorescence microscope.
Cell proliferation assay
For cell proliferation, MTT assay was performed using CellTiter reagent (Promega) according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-wells plate (5 replicates for each experimental condition) having a density of 8 × 103 cells/well in RPMI (-FBS) and treated with 4 µL of EVs, 0.1% DMSO or 15 µM sorafenib, all of which were in the range of 10^8 according to the NTA analysis. After 24 h viability was assessed with the addition of 10 µL/well of sterile CellTiter reagent (Promega). The plates were incubated at 37 °C for 2 h in a humidified, 5% CO2 atmosphere and the absorbance at 490 nm was recorded using the microplate reader EnSight (PerkinElmer, Waltham, MA). For assessing the viability, a 1:2 dilution of cell suspension was made in 0.4% trypan blue stain in a 0.2 ml tube (10 µl of cell suspension added to 10 µl of trypan blue). Ten microliters of the mixture were loaded into the opening of the TC20 counting slide (Bio-Rad). The slide was then inserted into the TC20 instrument (Bio-Rad) and cells counted within 10 min of trypan blue addition.
Zebrafish maintenance and eggs collection
Danio rerio (zebrafish) was maintained and used according to EU Directive 2010/63/EU for animal use following protocols approved by the local committee (OPBA) and authorized by the Ministry of Health (Authorization Number 287/2018). Fish were maintained in 3 L of water at a controlled- temperature (28.5 ℃) with 14 h light and 10 h dark cycle and fed 3 times per day, 2 times with dry food and 1 time with artemia [32]. Mating was set up in organized tanks, upon fertilization the eggs were collected and placed in a Petri dish containing fish water and incubated at 28 °C. Tricaine (MS222; E10521, Sigma–Aldrich St. Louis, MO, USA) was added to the fish water for zebrafish embryos and larvae anesthesia at 0.02% fnal concentration. The wild-type line used in this work included an AB strain (KIT Institute -Karlsruhe-Germany) and a transgenic line Tg(kdrl-EGFP) [33].
EVs uptake by immersion method
Stock solutions of EVs were prepared for embryo exposure at 5, 10, and 15 µl in 2 mL fish water. Embryos were collected, dead embryos were discarded, and alive embryos were transferred in new Petri-dish. At the gastrula stage (5 hpf) embryos were exposed to EVDMSO or EVSorafenib at the selected doses by the classic immersion method [34] up to 96 hpf. As a negative control, embryos were exposed to fish water plus 0.1% DSO (expected mortality rate < 15%). As a positive control for survival rate experiments, we used 3,4-dichloroaniline (DCA) (Sigma-Aldrich) dissolved in fish water at a concentration of 3.74 mg/L (expected mortality rate > 85–90%) [35]. For each treatment condition (EVDMSO and EVsorafenib), 30 embryos were used, and experiments were repeated three times. For all experiments only positive embryos (embryos labeled with marked EVs with DiI) were taken into consideration. The survival rate was recorded at 24, 48, 72 and 96 hpf respectively. The calculated percentages of dead embryos were below 15%, which is the expected mortality according to the OECD 2019 guidelines (Test Guideline No. 203, Fish Acute Toxicity Testing) [36] and a dose-response graph was plotted.
Induction of tumor xenografts
Tumor xenograft experiments were performed according to the protocol established by Ren J. et al. [37]. To evaluate the effect of EVs on tumor growth, 48 hpf Tg (kdrl:EGFP) zebrafish embryos were dechorionated, anesthetized with tricaine at 0.02% fnal concentration, followed by microinjection of the labeled MDA-MB-231 and MDA-MB-453 cells into the perivitelline space (PVS) [38]. Microinjections were performed with a FemtoJet electronic microinjector coupled with an InjectMan N12 manipulator (Eppendorf Italia, Milan, Italy). Approximately 250 cells/4 nL were injected into each embryo (about 25 embryos/group); embryos were maintained in PTU/fish water in a 32 °C incubator to allow tumor cell growth. Pictures of injected embryos were acquired using Zeiss Axiozoom V13 (Zeiss, Jena, Germany) fluorescence microscope, equipped with Zen pro software, 2 h after cell injection (T0). Using the same concentration range of 108 EVs, 15 µl of EVsorafenib or EVDMSO was added directly to the injected embryos in PTU/fish water. The calculated percentages of dead embryos were below 15% [36]. At 1-day post-injection (T1) and at 3-days post-injection (T3), the effects of the EVs-based treatment on tumor xenografts growth were scored by representative pictures, to measure the tumor areas of each group at T0, T1 and T3 using Zen Blue software from ZEISS. Embryos with micrometastases (indicated by the presence of at least one fluorescence dot outside the site of injection) and metastatic cells in the tails were counted and some representative xenografted embryos were fixed and embedded in low-melting agarose for image analysis.
Image acquisition and analysis
Bright-field and fluorescence images of embryos at different development stages (anesthetized with tricaine 0.16 mg/mL embedded in 0.8% low melting agarose and mounted on a depression slide) were captured using a Zeiss Axio Zoom V16 equipped with Zeiss Axiocam 506 color digital camera and processed using Zen 3.5 (Blue Version) software from Zeiss (Oberkochen, Germany), magnification 32x and 40x. Embryos were also observed using AxioObserver.Z1/7 with Apotome 3, Objective EC Plan-Neofluar 20x/0.50 (Carl Zeiss S.p.A., Milan Italy). For Light Sheet image acquisition embryos were first anesthetized using tricaine (0.02% in fish water) and subsequently included in a low melting agarose matrix (Top Vision Low Melting Point Agarose, Thermo FisherScientific) (0.5% in fish water). Images were acquired using Zeiss LightSheet microscope V1 supported by ZenPro software using a 488–30 nm laser and 505–545 nm filter. Images from the same experiment were taken with the same laser intensity and exposure time to generate comparable images. After the acquisition, 3D images were generated and manipulated using Arivis Vision 4D (Zeiss Oberkochen, Germany) 3D reconstructions of EGFP-positive cells were manipulated to obtain pictures comparable to each other in terms of fluorescence intensity. 3D reconstructions were exported as a single snap with the same compression setting.
Alkaline phosphatase (AP) assay
AP assay was performed according to Serbedzija et al. [39]. Briefly, embryos (n = 25) at 1-day post-injection (72 hpf) were fixed in 4% paraformaldehyde (PFA) and then put in 100% (v/v) methanol. The embryos were then equilibrated in Tris buffer (100 mM Tris HCl pH 9.5, 50 mM MgCl2, 100 mM NaCl, 0.1% Tween-20) and stained with nitro blue tetrazolium chloride (NBT) and 5-bromo-4-chloro-3’-indolyphosphate p-toluidine salt (BCIP) solution. The images were taken in a lateral and dorsal position at 32× magnification with a Zeiss AxiozoomV13 (Zeiss, Jena, Germany) microscope, equipped with a PlanNeoFluar Z 1×/0.25 FWD 56 mm lens and Zen Pro software.
Statistics and reproducibility
Statistical analysis was carried out using GraphPad Prism v8.0 (GraphPad Software, Inc., San Diego, CA, USA) software. Unpaired Student’s t-test was used to determine the differences of ncRNAs levels in EVs between the sorafenib treated cells and their control (0.1% DMSO) and for cell proliferation after the EV-based treatment. Analysis of variance (ANOVA) followed by post-hoc Tukey’s test was used to determine the significant differences in the ncRNAs levels after EV-based treatment as well as in the measurement of the tumor area, the assessment of micrometastases and angiogenesis in xenograft. All experiments conducted in vitro were performed two times, while in vivo experiments conducted in zebrafish, were performed three times and the number of embryos for each experimental point was n = 30. Data was considered statistically significant when the P-value ≤ 0.05.










留言 (0)