Culture and identification of hUC-MSCs
hUC-MSCs were procured from Pricella (CP-CL11, Wuhan, Hubei, China). hUC-MSCs were isolated and purified using the tissue explant method, and kept in specialized complete medium. Once the density reached 80%, the cells were disassociated using 0.25% trypsin (Pricella) and cultured for passage. The third- to fifth-passage cells were collected for subsequent analysis.Thereafter, their morphology was visualized through an inverted microscope (Olympus). Surface markers of hUC-MSCs (CD29, CD44, CD90, CD43, CD14 and HLA-DR) were detected using flow cytometry. The following flow cytometry antibodies (BD Biosciences, San Jose, CA, USA) were added at a concentration of approximately 1 µg/1 × 106 cells (optimal concentration was determined by the recommended concentration in the products’ instructions and by antibody titration experiments): purified anti-human CD29 antibody (303001), purified anti-human CD44 antibody (397502), purified anti-human CD90 (Thy1) antibody (328101), purified anti-human CD43 antibody (343202), FITC anti-human CD14 (982502) and purified anti-human HLA-DR antibody (327002). Cell purity of hUC-MSCs was determined using a flow cytometer (BD Biosciences) and analyzed by FlowJo V10 (FlowJo, BD Biosciences).
hUC-MSC-EVs isolation and identification
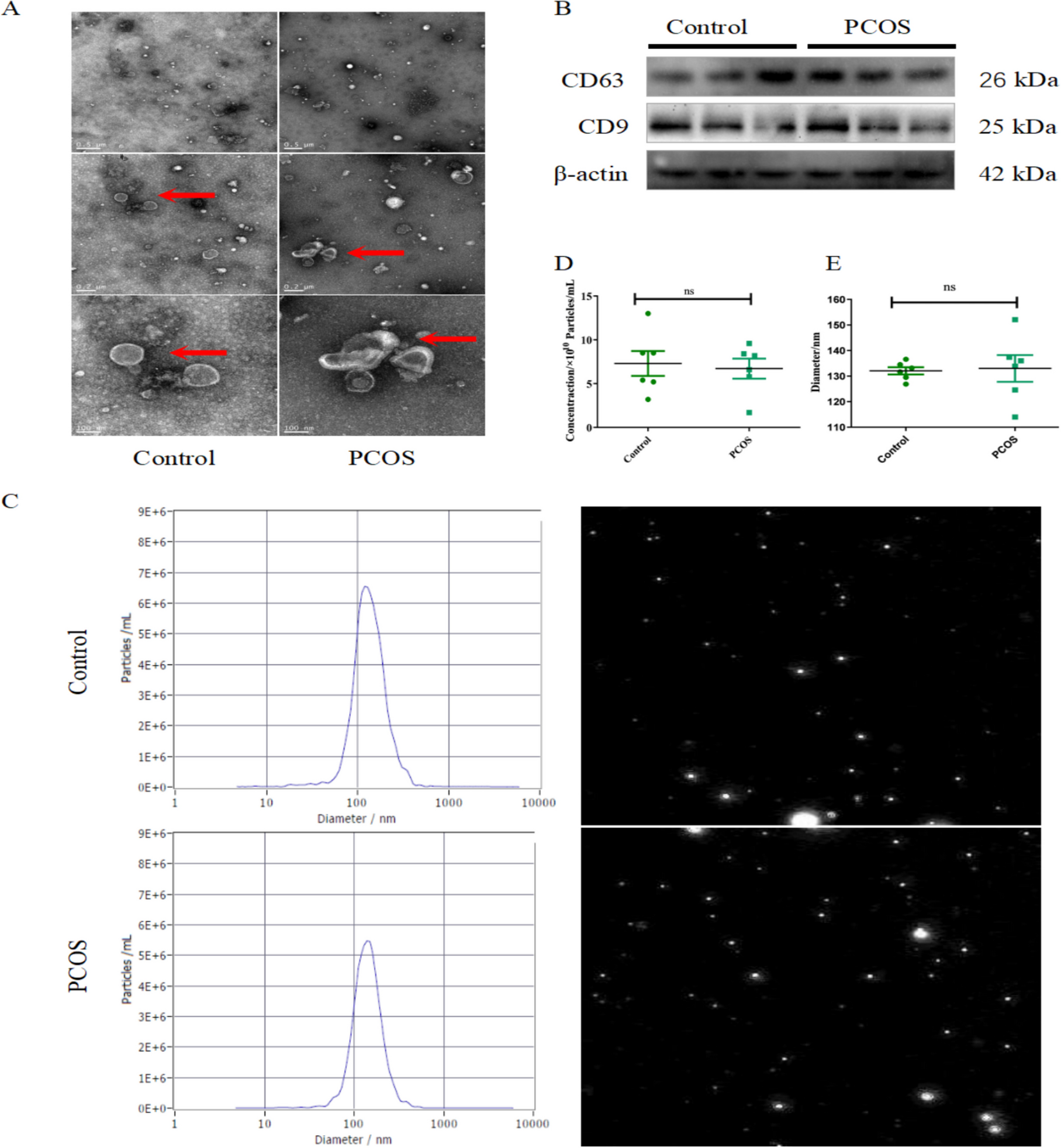
Supernatant collected from hUC-MSCs were centrifuged for 30 min at 2000×g and added with total exosome isolation reagent (Thermo Fisher Scientific, Waltham, MA, USA). After that, the supernatant was concentrated and placed at 2–8ºC overnight before centrifugation at 10,000×g and 4ºC for 1 h. Resuspended in 100 µL phosphate buffer solution (PBS), the collected EVs were observed with a transmission electronic microscope (TEM) (Olympus, Tokyo, Japan) and their diameter distribution and concentration were analyzed through nanoparticle tracking (NTA). Surface markers (CD9, CD63, CD81 and Calnexin) were quantified through western blot The supernatant of hUC-MSCs treated for 2 h with 20 µM GW4869 (inhibitor of EV secretion, Sigma Aldrich, St. Louis, MO, USA) [25] was served as the negative control (GW group).
EVs used in this study were divided into the following groups: (1) EVs: directly extracted from hUC-MSCs; (2) EVs + PK: hUC-MSCs-EVs treated with Proteinase K (20 mg/mL, Beyotime, Shanghai, China); (3) EVs + PK + T: hUC-MSCs-EVs treated with 20 mg/mL Proteinase K and 1% Triton-X-100 (Sigma Aldrich) [26]; (4) EVs-negative control of short hairpin RNA (sh-NC): EVs extracted from hUC-MSCs that were infected with blank lentiviral vector (pLenti-III-Blank Vector) (sh-NC); (5) EVs-sh-IGF-1: EVs extracted from hUC-MSCs that were infected with IGF-1 lentiviral vector (human; cytomegalovirus, pLenti-GIII-CMV) (sh-IGF-1). sh-IGF-1 and sh-NC were from Applied Biological Materials Inc. (Richmond, BC, Canada), with a titer of 1.5 × 108 TU/mL.
Animals
Specific pathogen-free 8-week-old female C57BL/6 mice [Shanghai SLAC Laboratory Animals Co., Ltd. (China)] were reared in standard animal rooms at temperature of 22–24 °C and humidity of 40–70% with 12 h/12 h light and dark cycles and free access to feed and water. All experiments were carried out after the review and approval from the ethics committee of Changzhi Medical College and conformed to internationally recognized animal research guidelines and ethics norms [Approval No. (2021)043].
Animal groups and treatment
Experimental POI was induced in mice by intraperitoneal injection of 120 mg/kg cyclophosphamide (CTX) once a week for 2 consecutive weeks [27], and mice administrated with normal saline at the equal amount and frequency served as the controls (control group).
According to the random number table, the mice were separated into the control, POI, POI + PBS, POI + EVs, POI + EVs-sh-NC, and POI + EVs-sh-IGF-1 groups (N = 6). In addition to intraperitoneal injection of 120 mg/kg CTX at the aforementioned amount and frequency, mice in the POI + PBS and POI + EVs groups were given 0.2 mL PBS or suspension encompassing 1 × 108/mL hUC-MSCs-EVs through their tail veins once every two days for 6 consecutive weeks [28], while mice in the POI + EVs-sh-NC and POI + EVs-sh-IGF-1 groups were administrated with 0.2 mL suspension comprising 1 × 108/mL EVs-sh-NC or EVs-sh-IGF-1 once every two days for 6 consecutive weeks.
One week after completion of all interventions, the mice were anesthetized using 0.3% pentobarbital sodium (50 mg/kg) intraperitoneally, and 0.1 mL of blood was sampled from the orbital venous plexus, with the serum separated for enzyme-linked immunosorbent assay (ELISA). Subsequently, the mice were euthanized by overdosing 3% pentobarbital sodium (100 mg/kg) intraperitoneally, and ovarian tissues were immediately collected. Part of the ovary was made into tissue sections for hematoxylin and eosin (H&E) staining and immunohistochemistry, and the others were made into tissue homogenates for western blot and detection of oxidative stress-related indexes.
ELISA
Mouse serum E2 (E-OSEL-M0008), follicle-stimulating hormone (FSH, E-EL-M0511), luteinizing hormone (LH, E-EL-M3053) and AMH (E-EL-M3015) levels were determined following the instructions of ELISA kits (all from Elabscience Biotechnology; Wuhan, China) and data were acquired with a microplate reader (Bio-Rad 680, Bio-Rad, Hercules, CA, USA).
H&E staining
Ovarian tissues were routinely fixed, dehydrated, embedded, and cut into 5 μm-thick sections. Staining was performed using the H&E kit (Solarbio Science & Technology Co., Ltd, Beijing, China) according to the manufacturer’s instructions. The pathological changes of mouse ovarian tissues were observed with an Olympus optical microscope, and the follicles observed were counted. The follicles were counted as hereinafter: the continuous cross-section slices of mouse ovarian tissues were collected. Subsequently, one every 12 slices were selected, with the numbers of primordial, primary, secondary, mature and atretic follicles documented, respectively. The morphological characteristics of the follicles at all levels were as follows: primordial follicles contained a layer of flat GCs surrounding the oocyte; primary follicles contained a layer of cubic GCs surrounding the oocyte; secondary follicles contained multiple layers of cubic GCs surrounding oocytes, without the formation of follicular lumen; mature follicles had the formation of a follicular lumen; and atretic follicles had the oocyte disappeared, with the GCs arranged irregularly and a pyknosis. Five sections from each mouse were taken for counting (6 mice/group). The results were signified as mean values.
Immunohistochemistry
After conventional dewaxing, rehydration, antigen recovery, and inactivation of endogenous peroxidase, the ovarian tissue sections were incubated with antibodies against IGF-1 (ab223567, 1:100, Abcam, Cambridge, MA, USA), FSH receptor (FSHR) (a specific marker for GCs) (ab113421, 1:100, Abcam), LC3 (ab192890, 1:2000, Abcam), and Beclin-1 (ab62557, 1:100, Abcam) at 4ºC overnight. The tissue sections were then washed and subject to incubation with goat anti-rabbit IgG H&L (horse radish peroxidase, HRP) (ab6721, 1:1000, Abcam) for 30 min. The nuclear was stained with diaminobenzidine (Sigma Aldrich), and sections were counterstained with hematoxylin. The sections were observed with an optical microscope (Olympus) and quantified with Image J software (National Institutes of Health, Bethesda, MD, USA).
In-vitro cell culture
Mouse ovarian GCs were obtained from Pricella (CP-M050, Pricella). The mouse ovarian tissues were mechanically separated, and then detached by collagenase, with the mouse GCs prepared by differential adhesion method. The GCs were kept in specialized complete medium (CM-M050, Pricella), and were detected for the GC-specific marker FSHR by immunofluorescence, which showed a cell purity of more than 98%, good cellular vitality, and free of HIV-1, HBV, HCV, mycoplasma, bacteria, yeast and fungi (Supplementary Fig. 1). The cultures were placed in a humidified environment at 37 °C with 5% CO2 and 95% air, and the medium was replaced every 2 days.
Treatment and groups of mouse GCs
Mouse GCs were divided into the blank, CTX (GCs treated with 30 µM CTX for 24 h) [27], CTX + EVs and CTX + PBS (GCs treated with 30 µM CTX and 30 µg/mL hUC-MSCs-EVs or equivalent PBS for 24 h) [29], CTX + EVs-sh-IGF-1 and CTX + EVs-sh-NC (GCs treated with 30 µM CTX and 30 µg/mL EVs-sh-IGF-1 or EVs-sh-NC for 24 h), as well as CTX + EVs + ML385 groups [GCs pre-treated with 10 µM ML385 (a Nrf2 inhibitor, MedChemExpress, Monmouth Junction, NJ, USA) for 2 h followed by treatment with 30 µM CTX and 30 µg/mL hUC-MSCs-EVs for 24 h, the concentration of ML385 was selected referring to a previous similar study amd product instructions [30]].
Uptake of EVs by GCs
EVs were labeled with PKH26 red fluorescent dye (MedChemExpress) and cultured with GCs for 24 h. GC slides were fixed in 4% paraformaldehyde (Beyotime) for 20 min, washed three times with PBS, stained with 4 -6-diamidino- 2-phenylindole (DAPI) (Beyotime) for 1 h, and the uptake of EVs by GCs was observed under a confocal fluorescence microscope (Carl Zeiss, Jena, Germany).
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
EVs and total cellular RNA were extracted by TRIzol regent (Invitrogen Inc., Carlsbad, CA, USA) and transcribed into cDNA with the PrimeScript RT kit (Takara Biotechnology Co., Ltd., Tokyo, Japan). PCR was conducted on an ABI 7900HT Rapid PCR Real-Time System (Applied Biosystems, Foster City, CA, USA) using SYBR® Premix Ex Taq™II (Takara) under the conditions of pre-denaturation at 95 °C for 10 min, denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s, and extension at 72 °C for 34 s for a total of 40 cycles. The experiment was repeated three times. Target gene expression was quantified using the 2−ΔΔCt method with β-actin as the housekeeping gene. See the primer sequences in Table 1.
Western blot
The hUC-MSCs, EVs, ovarian tissues and GCs were lysed on ice by RIPA lysis solution (Beyotime) to isolate nuclear and cytoplasmic proteins as per the instructions of the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime). Proteins were routinely isolated, transferred to a membrane, and blocked before incubation with primary antibodies against CD9 (ab236630, 1:1000, Abcam), CD63 (ab134045, 1:1000, Abcam), CD81 (ab109201, 1:1000, Abcam), Calnexin (ab22595, 1:1000, Abcam), IGF-1 (ab223567, 1:1000, Abcam), Nrf2 (GTX103322, 1:1000, GeneTex), HO-1 (GTX101147, 1:1000, GeneTex), LC3 (ab192890, 1:2000, Abcam), Beclin-1 (ab62557, 1:1000, Abcam), β-actin (ab8227, 1:2000, Abcam) and Lamin B1 (GTX103292, 1:1000, GeneTex) at 4 °C for 12 h. The membrane was washed in Tris-buffered saline with Tween (Solarbio) and incubated with goat anti-rabbit IgG H&L (HRP) (ab6721, 1:1000, Abcam) for 2 h at room temperature. Following development with ECL working solution (EMD Millipore, USA), the gray value of bands in the western blot images was quantified through ImageJ software (version 1.61; NIH Image), with β-actin as an internal control of cytoplasmic protein and Lamin B1 as an internal control of nuclear protein. Each experiment was repeated three times.
Cell counting kit-8 (CCK-8) for GC proliferation
GCs were seeded onto a 96-well plate with a density of 1 × 104 cells/well and cultured for 0, 6, 12, and 24 h to detect their proliferation ability with the CCK-8 kit (Beyotime).
Oxidative stress-related indicators
ROS (S0033M), glutathione (GSH; S0052) and malondialdehyde (MDA; S0131) contents in mouse ovarian tissues or GCs were determined as per the manufacturer’s instructions.
Lactate dehydrogenase (LDH) for GC death
Cell death was assessed by detecting LDH release using the LDH assay kit (Beyotime). The absorbance of the samples was measured at 490 nm using a microplate reader to calculate the amount of LDH release. LDH release (%) = (OD value of the experimental group - OD value of the blank group)/(OD value of the control group - OD value of the blank group) × 100%.
Immunofluorescence
Mouse GCs were fixed in PBS containing 4% paraformaldehyde for 20 min and permeabilized with 0.5% Triton X-100 for 15 min. After being blocking with 2% BSA, the GCs were incubated overnight in a humidified chamber at 4 °C with primary antibodies against Nrf2 (GTX103322, 1:100, GeneTex) and LC3 (ab192890, 1:100 Abcam). Subsequently, incubation with secondary antibody goat anti-rabbit IgG H&L (Alexa Fluor® 488) (ab150077, 1:200, Abcam) was performed for 2 h at room temperature. Nuclei were counterstained with DAPI (Beyotime) for 5 min. The stained sections were visualized with an Olympus BX53 microscope and quantified with ImageJ software (version 1.61; NIH Image) for co-localization.
Statistical analysis
Statistical analysis and graph drawing for all data were carried out by GraphPad Prism 8.01 (GraphPad Software Inc). Outlier detection was performed by Grubbs’ test and no data point was excluded by the test. Measurement data, expressed as mean ± standard deviation, were subject to independent t test in case of two groups, and one-way analysis of variance in case of multiple groups, followed by Tukey’s multiple comparisons test. Two-sided P value of < 0.05 was deeded to have statistical significance.









留言 (0)