
記住我






Six-week-old, sexually mature female albino rats weighing 180–200 g and in the diestrus (DE) phase were obtained from the Experimental Animal Unit of the Faculty of Veterinary Medicine, Benha University, Egypt, and were acclimatized for 2 weeks under standard laboratory conditions. Every day, vaginal smears were used to examine the various stages of the estrus cycle. Sterile cotton swabs dipped in phosphate buffer saline (PBS) were used to prepare the vaginal swabs, which were subsequently fixed in absolute methanol. Following fixation, the smears of swabs were stained with Giemsa, and assessed under a microscope (Nikon, Japan). Animals were bred and raised in pathogen-free cages and were exposed to room temperature (23 ± 3 °C) and a standard 12-hour light/dark cycle beginning at 8:00 AM. They were also allowed unrestricted access to clean water and food.
Source and localization of lyophilized MSCs-derived extracellular vesicles (MSCs-EVs)Lyophilized MSCs-EVs powder (Bioluga ® Canada) was reconstituted in distilled water, and each 1 ml of the MSCs-EVs was derived from 0.5 × 106 MSCs. Immunohistochemistry staining for CD105 was performed to localize administrated MSCs-EVs in ovarian tissues.
Ovarian failure (OF) inductionBusulfan (Sigma-Aldrich, Missouri, B2635, 10 mg/kg) was administrated to rats every day for 4 days, and for the first 2 days, they were given daily 100 mg/kg cyclophosphamide (Baxter, India) [24]. Histological and hormonal assessments were used to confirm OF after 4 weeks of induction. Dimethyl sulfoxide (DMSO, Sigma-Aldrich) was diluted (1:1) with sterile water and used to dissolve busulfan to be administered intraperitoneally (i.p.). One to two hours after the busulfan injection, cyclophosphamide dissolved in sterile injection-grade water was i.p. administered.
Experimental designThe timeline of treatments is indicated in Fig. 1. Fifty animals were randomly grouped into five groups as follows (n = 10):
Fig. 1
Schematic presentation of the timeline of treatments
Control groupAnimals received sterile injection grade water i.p. and DMSO diluted with water (1:1) for the first 2 days. Then, the diluted DMSO was continued for a further 2 days. Animals were left untreated for 4 weeks. After that, PBS was i.p. administrated for 6 weeks.
OF groupAfter 4 weeks of OF induction, rats were intraperitoneally injected with PBS for 6 weeks.
OF + MSCs-EVs groupMSCs-EVs were intraperitoneally injected (as a single dose) 1 week before induction of OF and then after 4 weeks of induction; 2 doses of MSCs-EVs, were intraperitoneally injected 2 weeks apart, then the experiment was ended 4 weeks after the second MSCs-EVs injection. For every animal, 0.5 ml of MSCs-EVs at a concentration of 100 µg protein/ml was administered [24]. The number and times of injections were selected based on previous studies [25] and preliminary experiments.
OF + rapamycin groupRapamycin (8 mg/kg) was administrated daily 1 week before OF induction. Then, 4 weeks after OF induction, rats were intraperitoneally injected daily for 6 weeks with rapamycin [15].
OF + quercetin groupRats were injected with daily Quercetin (40 mg/kg) 1 week before OF induction. Then, 4 weeks after OF induction, rats were intraperitoneally injected daily for 6 weeks with Quercetin [26].
It should be noted that the current study is limited by the lack of an MSCs-EVs-only treatment group to test the safety of MSCs-EVs and rule out any tumorigenic effects.
Sample collectionRats were fasted for 12 h at the end of the experimental period before receiving 10 mg/kg xylazine and 100 mg/kg ketamine by intramuscular injection to induce anesthesia. The blood was then withdrawn from the retro-orbital sinus using a capillary tube. Blood samples were used to prepare serum which was further used to assess gonadal hormones. Ovarian tissue samples were dissected and divided into three parts. The first part was fixed in formaldehyde 10% to be processed as paraffin blocks for histopathological and immunohistochemical studies. The second and third parts were processed to be used for biochemical and molecular analyses.
Gene expression profileTo extract total RNA from ovarian tissues, TRIzol (Invitrogen) was used according to the manufacturer’s instructions. For extraction and purification of miRNA, mirPremier microRNA isolation Kit (Sigma Aldrich, USA) was used. Using a Nano-Drop 2000 C spectrophotometer (Thermo Scientific, USA), the extracted RNA was tested for concentration and purity. A high level of RNA purity was considered at an A260/A280 absorbance ratio exceeding 1.9. The SensiFast cDNA synthesis kits (Sigma Bioline, UK) for RNA and the NCode VILO miRNA cDNA Synthesis Kit (Invitrogen, USA) for miRNA were used to synthesize complementary DNA (cDNA), following the manufacturer’s protocol. Next, quantitative PCR (qPCR) was performed using Maxima SYBR Green/ROX qPCR master mix (2x) (Thermo Scientific, USA). Table 1 indicates the sequence of primers used in this study. GAPDH and U6 were used for normalization. For the calculation of the relative gene expression ratios, the formula: RQ = 2−∆∆Ct was used [27].
Table 1 Sequences of primers for target genes and miRNA usedWestern blotOvarian tissue protein was extracted from different experimental groups. Laemmli buffer was added to protein samples followed by heating for 5 min at 95 °C. Then, 50 mg of protein samples were loaded onto sodium dodecyl sulfate (SDS, 10%) polyacrylamide gels followed by electrophoretic resolution. For blotting, the protein was then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Merk, Germany). The blots were blocked by 1-hour incubation in 5% nonfat dry milk in 0.1% TBS/Tween 20. Next, the blots were incubated with the appropriate primary antibodies for an entire night at 4 °C, PTEN (E-AB-63495, Elabscience, USA), FOXO3 (NBP2-16521, Novus Biologicals USA), mTOR (sc-517464, Santa Cruz Biotechnology, USA), Phospho-mTOR (sc-293133, Santa Cruz Biotechnology, USA), PI3K (E-AB-64202, Elabscience, USA), Phospho-PI3K (E-AB-20966, Elabscience, USA), AKT (E-AB-15441, Elabscience, USA), Phospho-AKT (E-AB-20804, Elabscience, USA), β-actin (E-AB-20031, Elabscience, USA). The blots were then washed before incubation with appropriate alkaline phosphatase (ALP) conjugated secondary antibody at room temperature for 1 h. For visualization of the protein bands, a BCIP/NPT detection kit (BWR1067, Biospes, China) was used, and for densitometric analysis, ImageJ® software was applied. To ensure normality, β-actin was employed as a housekeeping protein.
Enzyme-linked immunosorbent assay (ELISA) for AMH, FSH, LH, and E2Estradiol (E2, CSB-E05110r, Cusabio Biotech, USA), Follicle-Stimulating Hormone (FSH, CSB-E06869r, Cusabio Biotech, USA), Luteinizing Hormone (LH, CSB-E12654r, Cusabio Biotech, USA), and anti-Müllerian hormone (AMH, CSB-E11162r, Cusabio Biotech, USA) were measured in serum using commercially available ELISA kits in line with the guidelines provided by the manufacturer.
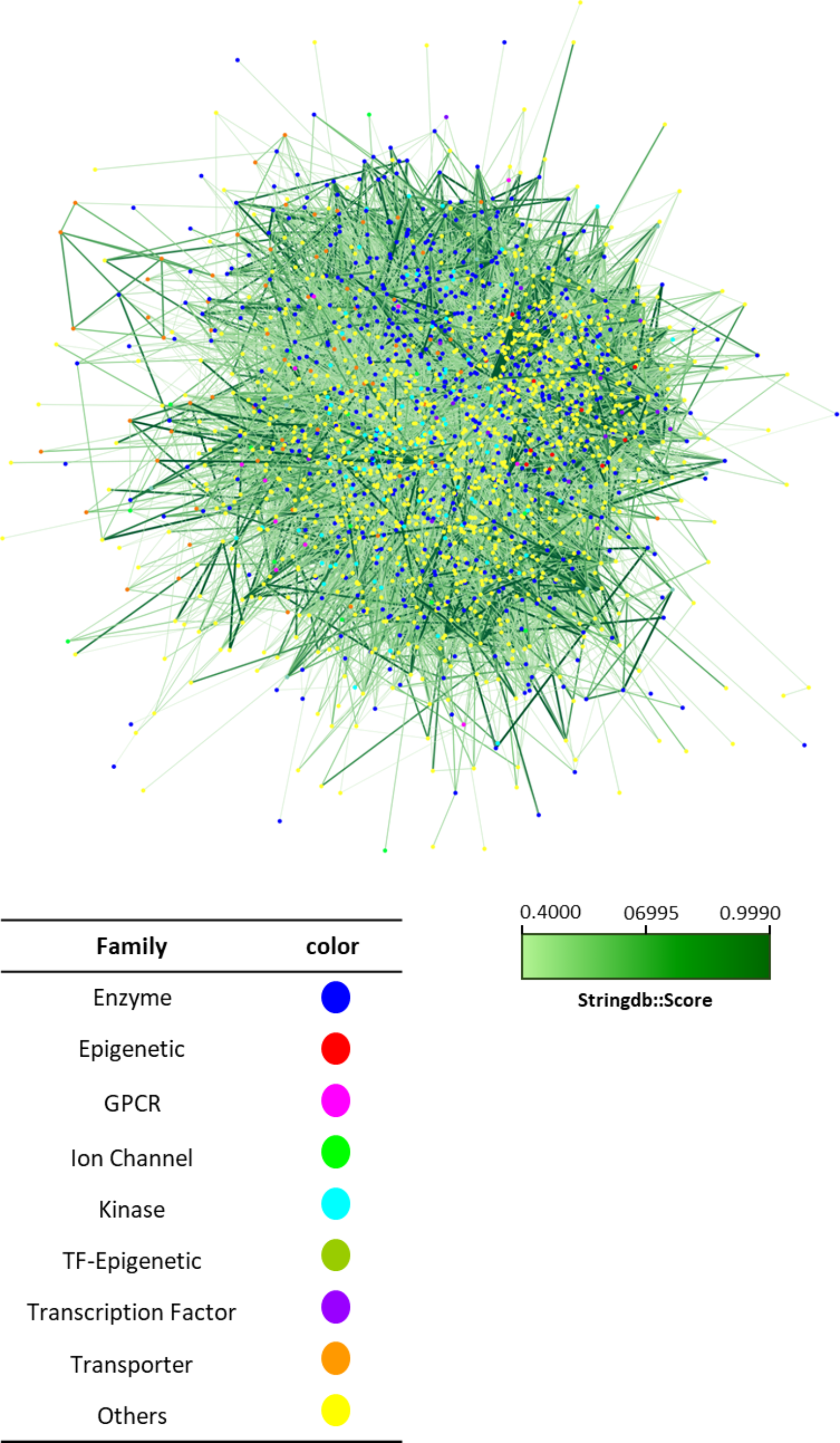
Analysis of molecular interaction network and targeted pathwaysThe analysis of the PTEN and FOXO3 genes was conducted through integrated transformation and correlation assessments using FunRich software (version 3.3; http://www.funrich.org/). This analysis aimed to elucidate molecular interaction networks and biological pathways, applying a statistical significance threshold of P < 0.05. Additionally, to evaluate the implication of specific exosomal miRNAs, including miR-200c, miR-122, and miR-99, FunRich was employed for molecular pathway enrichment analysis, again using the same P < 0.05 threshold. This comprehensive approach allowed for a detailed exploration of the functional roles of the studied genes and miRNAs in the context of ovarian toxicity and their potential therapeutic implications.
Histopathological analysisHaematoxylin and eosin (H&E) stainingOvarian sections of 4-6-µm-thickness were prepared from fixed ovarian tissue specimens. For the dehydration of fixed sections, ascending concentrations of ethanol were used. After two rounds of distilled water washing, the dehydrated sections were stained with H&E. Using a light microscope (Leica DMR 3000; Leica Microsystem), Ovarian tissue sections were inspected and analyzed, and images were captured by two experienced investigators who were blinded to the process [28].
Immunohistochemistry analysisDeparaffinized sections were hydrated, and then H2O2 (10%) was used to block endogenous peroxidase activity. The sections were then incubated with primary antibodies after being blocked for nonspecific reactions. The primary antibodies used were Anti-Ki67 antibody [SP6], (ab16667), Rabbit monoclonal [SP6] to Ki67, 1/200, Abcam), Anti-Bax antibody [E63] (ab32503), Rabbit monoclonal [E63] to Bax, 1/250, abcam, Anti-Bcl2-L-13 antibody [EP10625] (ab203516), Rabbit monoclonal [EP10625] to Bcl2-L-13, 1/500, Anti-CD105 antibody (Anti-CD105 antibody [8A1], ab230925) to CD105. Before incubating with the biotinylated secondary antibody, the slides were washed with PBS. After that, the slides were incubated with labeled avidin-biotin peroxidase, employing diaminobenzidine as a chromogen to visualize the antigen-antibody reaction.
Morphometric studyScoring of immunoreactivity was performed with the Allred score, which provides a 0–8 scale representing Allred index (0–1 = negative, 2–3 = mild, 4–6 = moderate, and 7–8 = strongly positive) [29]. Using the QuPath program (0.1.2), the percentage of positive cells and staining intensity grades (0–3) are added to determine the score [30]. Also, the number of primordial follicles (healthy and atretic) was assessed.
Statistical analysisGraphPad Prism, version 8 (GraphPad Software) was used for data analyses and presentation. Results were presented as mean ± standard error of mean (SEM). Statistical analysis was performed by applying one-way ANOVA test. Post-hoc analysis was then performed using Tukey’s test. The significance was considered at a P value less than 0.05.
留言 (0)