
記住我
As the largest microbial community in the human body, the gut microbiota plays a crucial role in establishing and maintaining host physiological homeostasis. A variety of human diseases are known to be associated with dysbiosis of the gut microbiota, such as obesity, cardiovascular disease and neurological disorders (Jin et al., 2019; Chen et al., 2021). The proposed theories of gut-liver axis, gut-brain axis, gut-lung axis, and gut-bone axis also fully illustrate the close relationship between the gut microbiota and various organs and systems in the human body (Cryan et al., 2019; Dang and Marsland, 2019; Albillos et al., 2020; Tu et al., 2021). The oral cavity is the second largest microbial habitat in the human body after the gastrointestinal tract (GIT). Studies have shown significant disease-specific patterns in the composition of the salivary microbiota of periodontitis patients (Lundmark et al., 2019). The dominant phyla included Firmicutes, Fusobacteria, Actinobacteria, Synergistes, Spirochetes, Proteobacteria, Saccharibacteria (TM7), and Bacteroidetes are highly associated with periodontal disease, contributing in part to the diversity and functional differences in the oral microbial community (Pei et al., 2020). The oral microbiota can be modified by systemic diseases with increased inflammation, such as diabetes mellitus (DM) and rheumatoid arthritis (RA), which consequently increase bacterial pathogenicity and susceptibility to periodontitis (Graves et al., 2019). Conversely, oral bacteria can impact systemic disease through bacteremia and has been linked to worsening of Alzheimer disease (AD), DM, cardiovascular disease and RA (Hajishengallis et al., 2022; Brewer et al., 2023). More interestingly, oral bacteria may indirectly affect these diseases by influencing the composition of the gut microbiota (Mesa et al., 2019; Sureda et al., 2020; Park et al., 2021; Barutta et al., 2022).
The oral cavity is directly connected to the GIT, and the progression of the ecological niche from the oral cavity to the gut has been defined as the “oral-gut microbiome axis” (hereafter referred to as the “oral-gut axis”). In addition to oral diseases, the oral microbiota is also involved in the regulation of extra-oral diseases through the oral-gut axis. The oral cavity is a microbial reservoir that constantly replenishes the gut microbiota (Schmidt et al., 2019). One study using 16S ribosomal RNA analysis provides evidence of widespread translocation of oral bacteria to the gut. After analyzing 144 pairs of saliva and stool samples, it was found that shared amplicon sequence variants between the salivary and gut microbiota were present in 72.9% of subjects, and that their total relative abundance in the gut was significantly higher in older subjects or those with dental plaque accumulation (Kurushima et al., 2023). Another large-scale controlled study of saliva and fecal samples taken separately from periodontally diseased/healthy subjects confirmed that periodontal status may indeed drive variations in the salivary and gut microbiota (Kurushima et al., 2023). Bao et al. further verified that periodontitis can induce intestinal dysbiosis and inflammation through the influx of salivary microbes by transplanting saliva from patients with periodontitis into mice via oral gavage (Bao et al., 2022). Furthermore, periodontal treatment may improve systemic health through the oral-gut axis further supports this concept and suggests target that periodontal therapy may be systemically useful by reducing the impact of the oral microbiota on intestinal bacteria. Analysis of stool and saliva samples from periodontitis patients using 16S ribosomal RNA gene amplicon sequencing confirmed that periodontal treatment both mitigated oral dysbiosis and altered gut microbial composition (Baima et al., 2024). Other evidence comes from studies examining a positive effect of periodontal therapy on the development of liver disease in cirrhotic patients by reducing bacterial dysbiosis in the feces (Bajaj et al., 2015; Bajaj et al., 2018). The need for a periodontal examination prior to liver transplantation has also been noted (Guggenheimer et al., 2007; Raghava et al., 2013).
How the oral and gut microbiomes interdependently regulate physiological functions to impact systemic health has not been fully investigated. In this paper, we provide potential mechanisms by which periodontal bacteria regulate extra-oral diseases via the oral-gut axis, and the far-reaching implications of maintaining periodontal health in reducing the risk of many intestinal and parenteral diseases.
2 Occurrence of periodontal pathogenic bacteriaHost–microbiome homeostasis in the oral cavity can be seen as an ‘armed peace’ that maintains a controlled state of inflammation. The transition from health to disease requires a susceptible host and a dysregulated oral microbiota. For susceptible host, as shown in diabetic mice, diabetes-enhanced IL-17 alters the oral microbiota and renders it more pathogenic (Xiao et al., 2017). On the contrary, local factors, such as poor oral hygiene, may induce oral microbial dysbiosis (e.g. altered proportions of coccobacilli), which ultimately leads to inflammation of the periodontal tissues (Lin et al., 2021). The ensuing increased flow of gingival crevicular fluid not only introduces a component of the host’s defenses, but also provides the substrate necessary for the growth of many ‘inflammophilic’ bacteria (i.e. those that thrive within an inflammatory environment, e.g. Porphyromonas gingivalis) (Hajishengallis, 2014). Inflammophilic bacteria actually gain a growth advantage over commensal microbiome in response to inflammation. With the aim of self-feeding, Inflammophilic bacteria develop a range of pathogenic strategies (e.g. affecting neutrophil functions) that gradually disrupt the ecological balance of the existing subgingival microbial community, leading to a more intense host response (Higashi et al., 2024).
The emergence of periodontal bacterial pathogenicity may be related to plaque biofilms attached to the surfaces of teeth, interdental spaces or restorations. Dental plaque represents the microbial community whose collective properties differ significantly from planktonic bacteria, which enhance the survival, metabolism and pathogenesis of oral microorganisms (Freire et al., 2021). Periodontitis-specific pathogenic bacteria, such as P.gingivalis, Treponema denticola, Filifactor alocis, Tannerella forsythia and Aggregatibacter actinomycetemcomitans, colonize and proliferate predominantly within the host periodontal pocket (Darveau et al., 2012). Three key factors may be involved in the induction of inflammation by periodontal pathogenic bacteria to promote periodontal tissue damage, including oral mucosal inflammation and alveolar bone destruction. The first is a change in the relative abundance of dominant species (e.g., inflammophilic bacteria). High levels of the genera Porphyromonas (32.2%), Fretibacterium (10.4%), Rothia (5.3%), and Filifactor (3.1%) were observed in periodontitis (Abusleme et al., 2021). Studies have shown that a very small proportion of Porphyromonas gingivalis in the community can orchestrate the normal benign microbiota into a dysbiotic community structure (Darveau et al., 2012). The second is related to the location of the plaque biofilm. In line with this view, Carrouel et al. noted that even in periodontally healthy young adults, the interdental space favors the development of periodontal disease as an ecological niche where microbial communities congregate (Carrouel et al., 2016). Compared to supragingival biofilms of other oral mucosa, the interdental biofilm is located between two teeth and the gingiva, where bacteria are in a more anaerobic environment. Due to its unusual anatomy, the body has few or no alternative defenses against the interdental space, and traditional methods of daily control (e.g. toothbrushing and saliva) are inadequate for biofilm disruption at this anatomical location (Carrouel et al., 2016). Quantitative real-time PCR assays have been applied to reflect microbial succession events in developing interdental biofilms, strongly suggesting that oral microbial dysbiosis is associated with the risk of periodontal and related diseases (Carrouel et al., 2016). The final factor is bacterial virulence. For example, gingipains are critical virulence factors for Porphyromonas gingivalis to colonize and proliferate in the gingival crevice and to invade the periodontium (Silva and Cascales, 2021). F. nucleatum mediates important biofilm-organizing behaviour and interactions with host cells through the expression of numerous adhesins (e.g. RadD) (Brennan and Garrett, 2019).
Because of the deleterious effects of periodontal pathogenic bacteria, there is increasing interest in whether daily antimicrobial strategies can prevent or reverse oral dysbiosis by altering the structure and function of the plaque biofilm, including healthy diet, oral hygiene, and the use of antimicrobials and probiotics. There have been studies showing changes in the oral microbiota associated with different dietary patterns, such as the amount of fermentable carbohydrates, fats, and anti-inflammatory/pro-inflammatory components, the degree of processing, and supplementation with nitrate (Anderson et al., 2020; Stanisic et al., 2021). An oral health optimized diet (low in carbohydrates, rich in Omega-3 fatty acids, and rich in vitamins C and D, antioxidants and fiber) has been found to reduce the load of potential cariogenic and periodontal bacterial species in the plaque biofilm, and even to reduce gingival and periodontal inflammation in humans (Tennert et al., 2020). However, some human studies have observed no clear relationship between diet and the composition of oral bacterial communities (Santonocito et al., 2022). Scholars holding this view believe that food is actually present in the mouth for a limited period of time and that the primary nutritional sources for oral bacteria appears to be saliva and gingival crevicular fluid. It has also been suggested that some dietary effects on the oral microbiota may occur indirectly by altering the gut microbiota. In conclusion, more research is still needed to elucidate whether healthy dietary patterns can prevent/reverse oral dysbiosis by altering the composition of oral microbiota/plaque biofilms.
In oral hygiene, the use of toothbrushes, interdental brushes, chewing gum, and even anti-plaque agents (e.g. chlorhexidine) are effective in reducing plaque or interrupting plaque maturation, but none of them seem to be able to influence the structure of the bacterial community in plaque. The main reason for this is the blocking effect of biofilms (Rosier et al., 2018). For example, although an anti-plaque agent blocks the formation of plaque, it has little activity against established plaque. The use of probiotics has the potential to prevent and/or treat oral and related diseases by reversing oral dysbiosis. Studies have shown that the addition of probiotics (e.g. Lactobacillus rhamnosus) does result in significant changes in the composition of oral bacterial communities, including an increase in microbial diversity and a decrease in the relative abundance of opportunistic pathogens (Di Stefano et al., 2023). It should be noted that this effect may be reversed after a period of cessation of treatment.
3 Possible pathogenic mechanisms of periodontal bacteria through the oral-gut axisMicrobial transmission between the oral cavity and the gut can shape and/or reshape the microbial ecosystem in both habitats, which in turn affects the overall microbial community of the organism. Multiple changes in the gut microbiota, intestinal barrier function and immune system induced by periodontal bacteria can lead to an increased risk of systemic diseases by promoting low-grade inflammation (Figure 1). Therefore, exploring the pathogenic role of periodontal bacteria through the oral-gut axis may help us to understand one of several avenues by which periodontitis may affect systemic diseases.
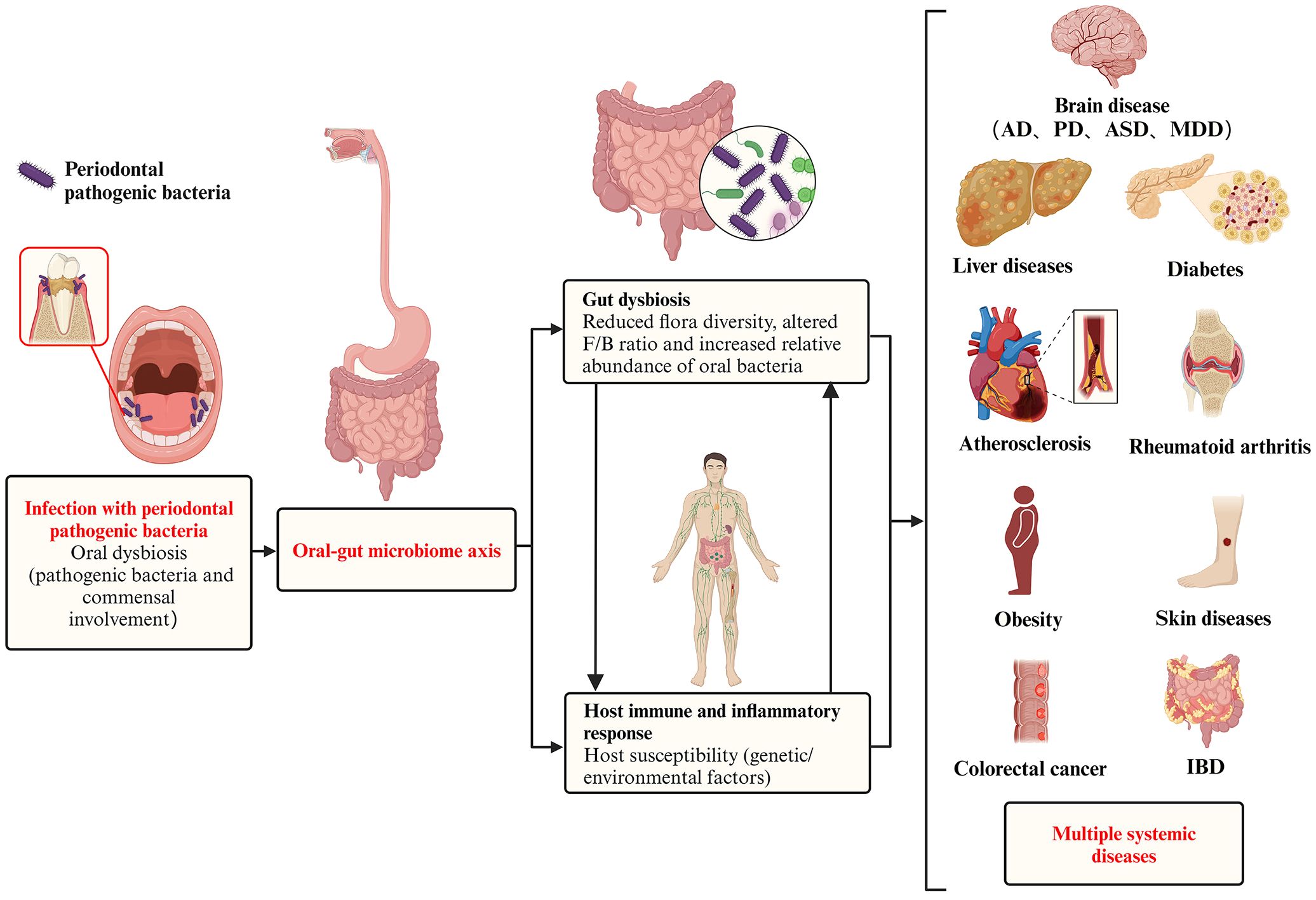
Figure 1. Pathogenic effect of periodontal pathogenic bacteria on systemic diseases via the oral-gut axis. In a pathogenic environment with excessive plaque accumulation, the dynamic equilibrium between microbial invasion and host defense is disrupted, and the periodontal pathogenic bacteria and commensal bacteria together give rise to oral dysbiosis and disease states. The oral cavity is directly connected to the gastrointestinal tract, and the progression of the ecological niche from the oral cavity to the gut has been defined as the ‘oral-gut microbiome axes. These periodontal pathogenic bacteria and their virulence products then breakthrough the barrier between the oral cavity and the gut and translocate in large numbers into the gut, causing gut dysbiosis, which is characterized by (i) a reduction in overall microbial community diversity, (ii) an alteration of the Firmicutes/Bacteroidetes (F/B) ratio, and (iii) as well as a reduction in probiotics and a corresponding increase in opportunistic pathogens. In addition to microbiota dysbiosis, the transition from health to disease requires a susceptible host (including genetic/environmental factors), accompanied by a complex set of interacting mechanisms that ultimately trigger destructive immune and inflammatory responses in the host. In addition to oral diseases, systemic diseases in which the oral microbiota may be involved in regulation via the oral-gut axis include intestinal diseases (e.g. inflammatory bowel disease and colorectal cancer), rheumatoid arthritis, brain diseases (e.g. Alzheimer’s disease, Parkinson’s disease, autism spectrum disorders, and major depressive disorder), liver diseases (e.g. non-alcoholic fatty liver disease, cirrhosis, and hepatocellular carcinoma), obesity, diabetes, atherosclerosis, and skin diseases.
The nature of the dysbiotic change that induces periodontitis has not been well defined and it has been difficult to distinguish between pathogenic and commensal bacteria. However, in the gut, dysbiosis has been identified as a reduction in alpha and beta diversity and an increase in bacterial pathogens, including a decrease in microbial population and functional diversity and stability (e.g. specific alterations in Peptococcaceae and Prevotellaceae), altered Firmicutes/Bacteroidetes (F/B) ratios, decreased abundance of beneficial SCFA-producing bacteria (e.g. Blautia, Roseburia, and Lachnospiraceae), and a corresponding increase in opportunistic pathogens (e.g. Megasphaera, Enterobacter, and Desulfovibrio) (Sultan et al., 2021; Chidambaram et al., 2022).
The gut originally is colonized by bacteria from the oral cavity or transit through the oral cavity. For example, Streptococcus salivarius and Streptococcus parasanguinis localized in the oral cavity most often colonize the intestinal niche (Kageyama et al., 2023). In addition, oral bacteria can alter the composition of the gut bacteria either directly or indirectly. In the intestine they may alter commensal bacteria through bacteria-bacteria interactions. For example, potential pathogens entering the gut can signal through quorum sensing to commensals and trigger the expression of toxins, virulence factors, and biofilm formation (Coquant et al., 2021). Periodontal bacteria (e.g. P. gingivalis) have been found to express a interspecies quorum sensing signal known as autoinducer-2 (AI-2), which modulates gut microbiota composition by enhancing Firmicutes growth and increasing the F/B ratio (Thompson et al., 2015; du Teil Espina et al., 2019).
A recent mouse study not only demonstrated that oral administration of P. gingivalis induced intestinal dysbiosis, reduced intestinal barrier function, and intestinal inflammation, but also further elucidated the pathological mechanisms behind the disruption of intestinal homeostasis by P. gingivalis through gut microbiota transplantation (Sohn et al., 2022). This study collected the gut microbiota of normal mice after oral administration of P. gingivalis and transferred it to genetically susceptible mice. The same intestinal inflammation was eventually observed in the recipient mice, verifying that P. gingivalis-induced intestinal dysbiosis is itself sufficient to promote intestinal inflammation (Figure 2).
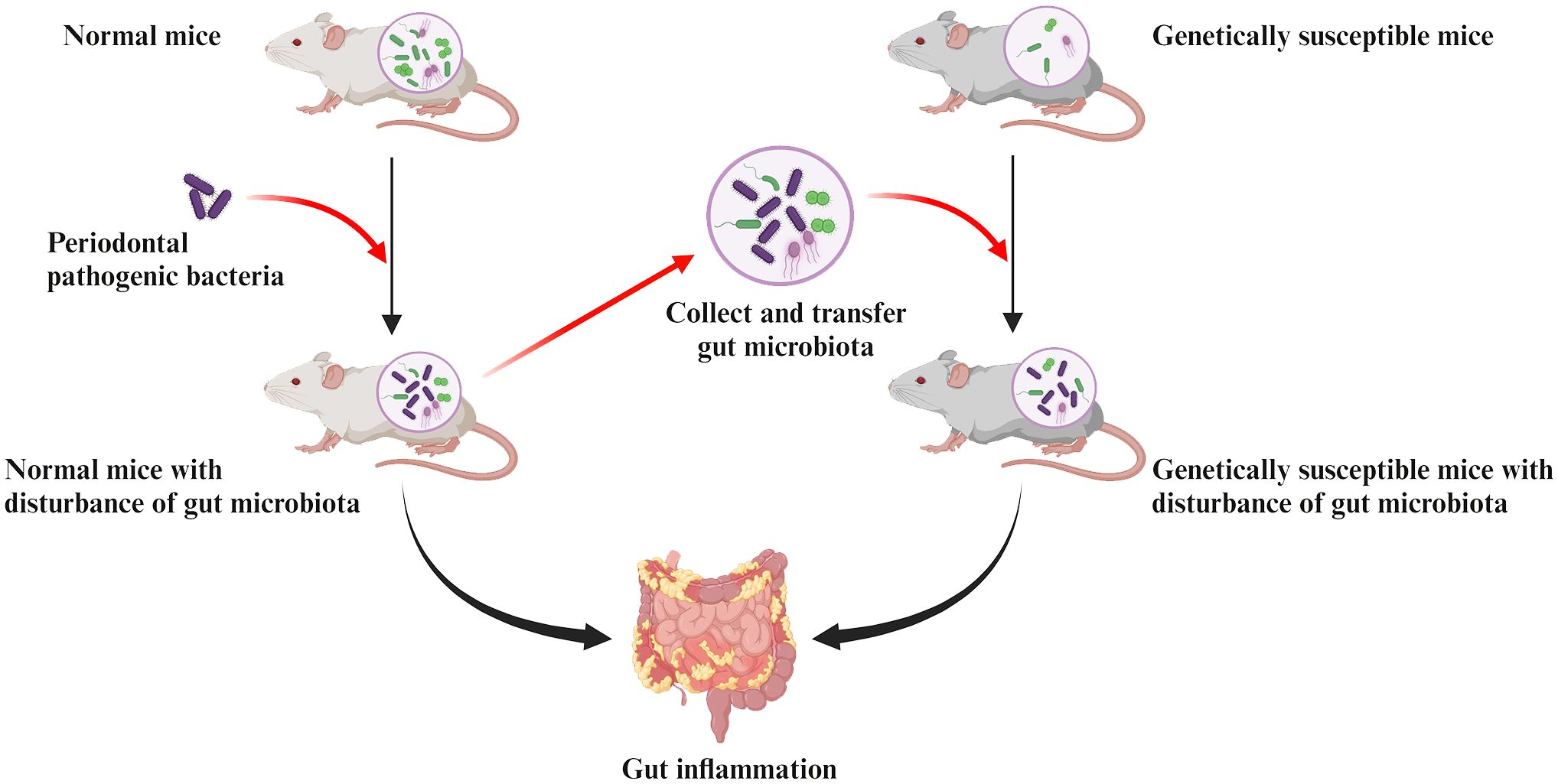
Figure 2. Pathologic mechanisms of intestinal inflammation induced by periodontal pathogenic bacteria are associated with the gut dysbiosis. Collecting the gut microbiota of normal mice after oral administration of periodontal pathogens and transferring it to genetically susceptible mice can observe the same intestinal inflammation in recipient mice. This confirms that the disruption of gut microbiota induced by periodontal pathogens is sufficient to cause their own intestinal inflammation.
3.1 Pathways of translocation of oral bacteria to the gutMore than half of the bacterial species in the gastrointestinal system undergo oral-gut translocation (Schmidt et al., 2019). First, the oral cavity and gut are inherently linked through saliva. Studies have demonstrated that periodontal bacteria migrating via enteral dissemination (salivary pathway) are able to colonize and survive in the gut for at least 24 hours (Bao et al., 2022). Second, the rich blood circulation in the oral cavity and the ulcerated surface of the lining of periodontal pockets in patients with periodontitis also allow the oral microbiota to colonize the gut through hematogenous dissemination. A study comparing colorectal cancer (CRC) colonization by gavage vs. intravenous inoculated Fusobacterium nucleatum in a mouse model and found that hematogenous fusobacteria were more successful in CRC colonization than gavaged ones. This may indicate that the circulatory system appears to be theefficient route for some periodontal bacteria to reach the gut. However, more evidence is still needed to support this.
Finally, after invasion of host cells, periodontal pathogenic bacteria (e.g. P. gingivalis and F. nucleatum) may survive within them and subsequently disseminate to the gut (Xue et al., 2018; Kitamoto and Kamada, 2022a). For example, periodontal bacteria with virulence factors that inhibit phagolysosome formation are able to survive within the host cell and migrate intracellularly via Trojan horse mode or vesicular trafficking (Yilmaz et al., 2004; Sansores-España et al., 2021). P. gingivalis has been shown to survive within macrophages, epithelial cells, endothelial cells and smooth muscle cells and to spread from one cell to another (Wang et al., 2007; Li et al., 2008; Carrion et al., 2012). Thus, theoretically, periodontal bacteria may hijack these cells as a vehicle for migration to the gut (Kitamoto et al., 2020; Hajishengallis and Chavakis, 2021). However, more evidence is still required to support that periodontal bacteria translocated via this pathway are of sufficient pathogenic significance in the oral-gut axis mediating systemic disease progression. In conclusion, enteral dissemination (the main pathway), hematogenous dissemination, and host cell hijacking are all possible ways in which the oral microbiota can translocate to the gut, resulting in a complex association of the oral-gut axis based on microbial crosstalk (Figure 3).
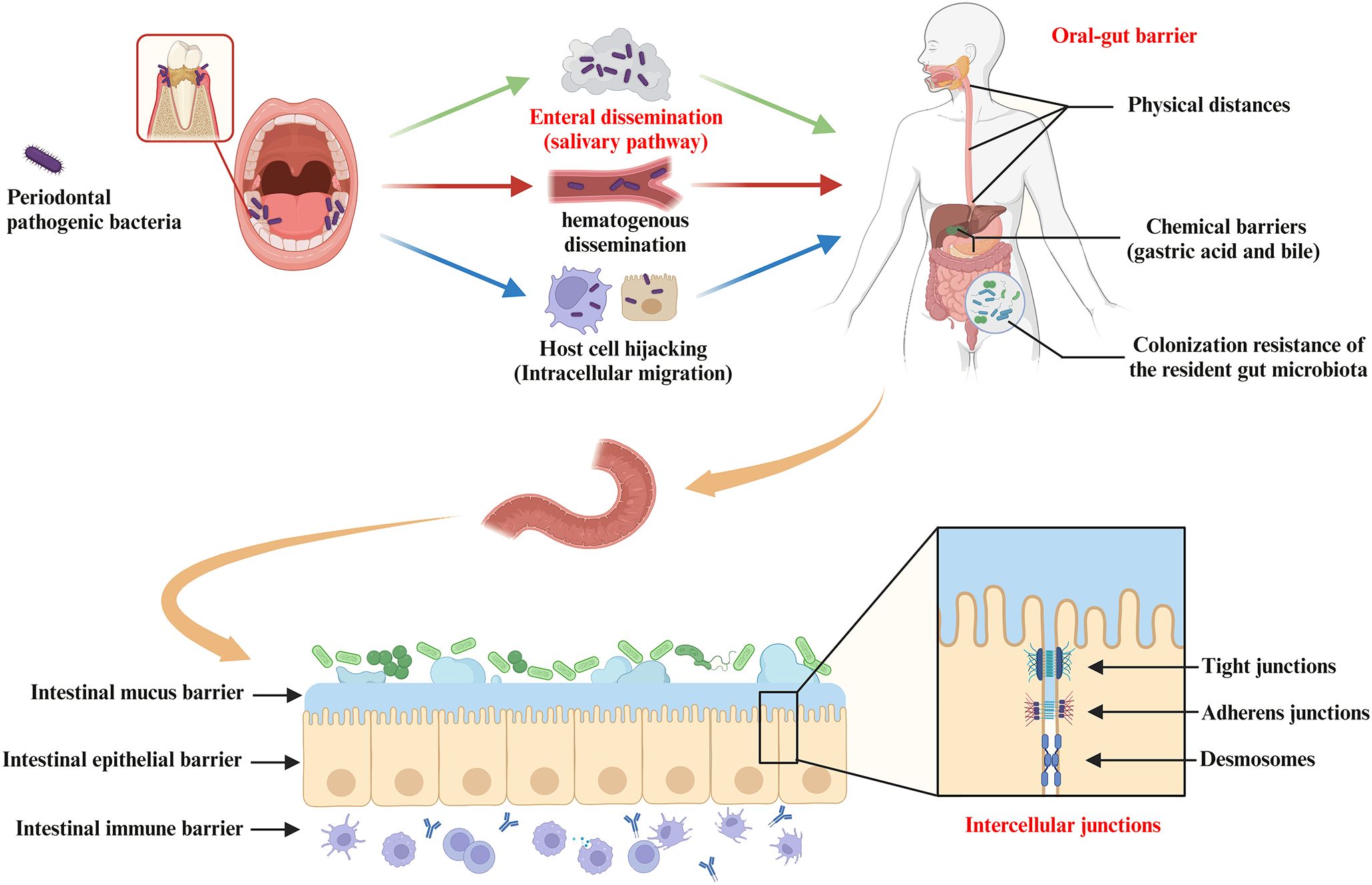
Figure 3. Pathways of oral bacterial translocation to the gut and host barrier systems limiting oral-gut microbial translocation. Intestinal translocation of periodontal pathogenic bacteria is pathogenically important in the progression of systemic diseases mediated by the oral-gut axis. Possible routes of oral microbiota translocation to the gut include (i) migration by enteral dissemination (salivary route), (ii) colonization of the gut by hematogenous dissemination, and (iii) hijacking of host cells as a vehicle for migration to the gut. First, the oral-gut barrier is one of the important strategies for the host to maintain microbial homeostasis in order to limit microbial translocation between the oral cavity and the gut. The oral-gut barrier includes physical distances and chemical barriers (e.g. gastric acid and bile), as well as colonization resistance of the resident gut microbiota to the intestinal migration of oral microbes with pathogenic potential. If oral bacteria break through the oral-gut barrier to reach the lumen, the barrier function of the intestinal wall is still able to separate the host from adjacent microbiota. The intestinal barrier consists of (i) the intestinal mucus barrier composed of mucin and antimicrobial peptides (AMPs) overlying IEC, (ii) the intestinal epithelial barrier composed of IEC and intercellular junctions (e.g. tight junctions, adherens junctions and desmosomes), and (iii) the intestinal immune barrier composed of innate and adaptive immune cells (gut-associated lymphoid tissue).
3.2 Breach of the oral-gut barrier by periodontal bacteriaThe oral-gut barrier is one of the important strategies for the host to maintain microbial homeostasis in order to limit microbial translocation between the oral cavity and the gut. The oral-gut barrier includes physical distances and chemical barriers (e.g. gastric acid and bile), as well as colonization resistance of the resident gut microbiota to the intestinal migration of oral microbes with pathogenic potential (Figure 3). Intestinal commensal bacteria can play an important role in regulating intestinal mucosal homeostasis and inhibiting colonization of potential oral pathogens via distinct mechanisms (Suárez et al., 2021). For example, intestinal commensal bacteria can compete for nutrients and produce antimicrobial peptides and metabolites that affect the colonization and virulence of oral pathogens. Intestinal commensal bacteria also promote the induction of effector T and B cells locally and systemically in response to pathogens (Suárez et al., 2021). However, this protective mechanism of colonization resistance may be disturbed by microbiota imbalance or local changes in host response, leading to disruption of host-microbiome homeostasis.
3.3 Effect of periodontal bacteria on intestinal barrier functionThe intestinal barrier has several physical and chemical barriers separating the host from the adjacent microbiota (Figure 3). However, when the intestinal barrier function is pathologically altered, increased intestinal permeability and inflammatory response may allow the gut to “leak”, i.e. allowing pathogenic substances to cross the intestinal wall and spread systemically with pathological consequences (Hollander and Kaunitz, 2020). Prolonged translocation of periodontal bacteria to the gut may further trigger and exacerbate the putative disease “leaky gut syndrome” by affecting gut microbiota homeostasis and barrier function, thus participating in the pathogenesis of various gastrointestinal and systemic diseases (Kinashi and Hase, 2021) (Figure 4). This will be discussed in more detail below.
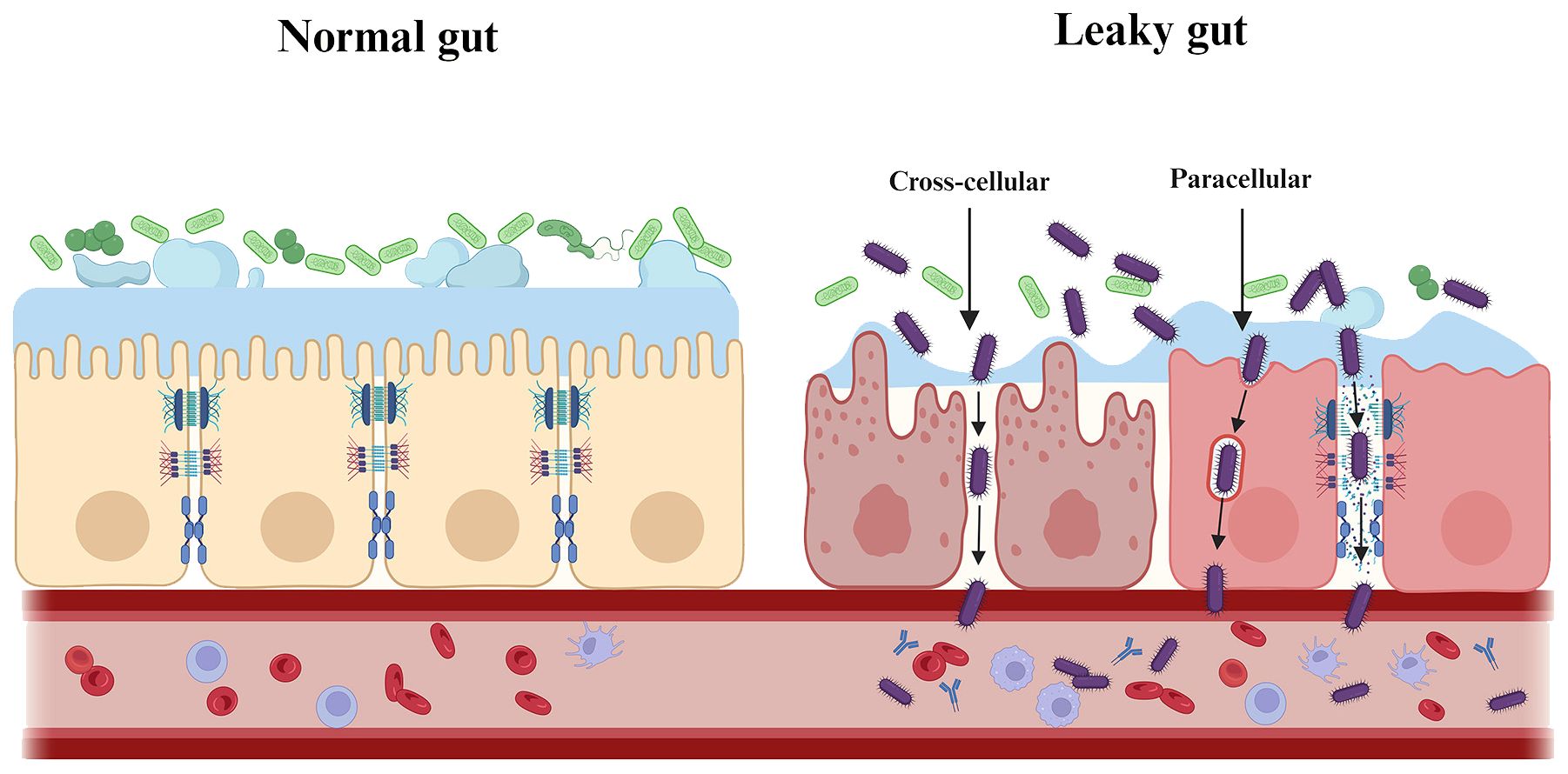
Figure 4. Periodontal pathogenic bacteria trigger and exacerbate “leaky gut syndrome”. Prolonged translocation of periodontal bacteria to the gut may further trigger and exacerbate the putative disease “leaky gut syndrome” by affecting gut microbiota homeostasis and barrier function. As shown, periodontal pathogenic bacteria are involved in the pathogenesis of various systemic diseases by either directly/indirectly reducing intestinal epithelial intercellular junctions (paracellular) or by invading intestinal epithelial cells (transcellular), which ultimately affects and breaches the intestinal barrier.
3.3.1 Intestinal mucus barrierA gel-like sieve structure formed by mucin overlying intestinal epithelial cells (IECs) is the first physical barrier that separates bacteria in the lumen from the IEC (Pelaseyed et al., 2014). At the same time, antimicrobial peptides (AMPs) (e.g. alpha-defensins, lysozyme C and C-type lectins), which are components of the intestinal mucus, form a chemical barrier that prevents and clears intestinal pathogens and protects intestinal cells from external factors (Birchenough et al., 2015; Liu et al., 2021b).
Periodontal bacteria are capable of disrupting the integrity and function of the intestinal mucus barrier. For example, P. gingivalis secretes Gingipain B (RgpB) that cleaves mucin 2 (MUC2) to disrupt its polymerization (van der Post et al., 2013). P. gingivalis can also circumvent or manipulate AMP to disrupt intestinal homeostasis, further exacerbating intestinal ‘leakiness’ (Ji et al., 2007; Hussain et al., 2015).
3.3.2 Intestinal epithelial barrierThe intestinal epithelial barrier is provided by IEC that form multiple types of junctions, including tight junctions (TJs), adherens junctions and desmosomes (Figure 3). The composition and abundance of the different components of intercellular junctions are decisive for intestinal permeability and, together with IEC, maintain an important mechanical barrier preventing the transit (cross-cellular/paracellular) of pathogenic substances such as toxins and bacteria from the lumen to other parts of the body. In addition, IEC can regulate the persistence of antigens on the surface through epithelial shedding (Suárez et al., 2021).
In affecting and breaching the intestinal epithelial barrier, periodontal pathogenic bacteria can directly/indirectly reduce inter-epithelial adhesions, or invade and transit apically through epithelial cells in several ways. 1) Downregulation of adhesion molecule expression. Pathogenic bacteria (e.g. P. gingivalis, A. actinomycetemcomitans and T. denticola) reduce intestinal epithelial surface expression of E-cadherin, resulting in the destruction of adherens junctions and allowing transmigration (Devaux et al., 2019). In animal studies, a downregulation of tight junctions at the mRNA level was also observed in the gut of mice following oral administration of periodontal pathogenic bacteria (e.g. P. gingivalis and F. nucleatum) (Meilian et al., 2016; Olsen and Yamazaki, 2019; Cao et al., 2020). 2) Degradation of adhesion molecules. For example, P. gingivalis uses gingival proteases to directly degrade tight junctions (e.g. Occludin) and adherens junctions (E-cadherin) (Takahashi et al., 2019; Tsuzuno et al., 2021). 3) Cause re-distribution of E-cadherin from membrane to cytoplasm. Substantial remodeling of cell junctions may attenuate the integrity of the intestinal epithelial barrier. The recombinant cyto-lethal distending toxin (Cdt), a putative virulence factor of A. actinomycetemcomitans, affects epithelial barrier function by altering E-cadherin’s cytosolic distribution (Takahashi et al., 2019). 4) Excessive cell death may lead to barrier dysfunction and translocation of pathogenic bacteria (Crawford et al., 2022). F. nucleatum and its LPS have been shown to promote cell apoptosis and pro-inflammatory cytokine production in gut by activating autophagy pathway in IEC in vivo and in vitro (Su et al., 2020).
Finally, periodontal pathogenic bacteria indirectly reduce inter-epithelial adhesion by stimulation of inflammation. Mechanistically, F. nucleatum targets caspase activation and caspase recruitment domain 3 (CARD3) to activate the endoplasmic reticulum stress (ERS) pathway or the IL-17F/NF-κB pathway, mediating damage to the intestinal epithelial barrier (Cao et al., 2020; Chen et al., 2020a). F. nucleatum can also increase inflammatory genes (e.g. NF-κB) through its FadA adhesin binding to E-cadherin (Pignatelli et al., 2023). In addition, studies have shown that even a single oral dose of P. gingivalis can cause the prevalence of inflammatory microbiota in the gut (Nakajima et al., 2015). The increase in the proportion of intestinal pathogenic bacteria leads to the enrichment of LPS in the lumen. LPS is an important stimulus for impaired intestinal epithelial barrier function via an intracellular mechanism involving TLR-4-dependent up-regulation of CD14 membrane expression (Ciesielska et al., 2021).
3.3.3 Intestinal immune barrierAs the largest immune organ in the body, the intestinal immune barrier consists of innate and adaptive immune cells (gut-associated lymphoid tissue). Disruption of the homeostasis of the gut microbiota and immune barrier may trigger an excessive intestinal immune response, leading to the intestinal barrier damage and ultimately inducing systemic diseases associated with altered intestinal immune responses (Liu et al., 2021b).
Studies have observed an upregulation of pro-inflammatory cytokines in the gut by periodontal pathogenic bacteria. Colonization by P. gingivalis drives an increase in pro-inflammatory factors such as IL-6 and TNF-α in the gut (Lee et al., 2022). P. gingivalis also induces an increase in intestinal-derived IL-17 in peripheral blood (leaky gut) and has been shown to be associated with increased LPS in the gut (Arimatsu et al., 2014; Sato et al., 2017). The exact mechanism of action currently requires further research to elucidate.
Disruption of the Th17/Treg balance and increased secretion of pro-inflammatory cytokines in the body are important ways in which periodontal pathogenic bacteria can trigger systemic disease by affecting immune homeostasis in the gut. Periodontal pathogenic bacteria in the gut have been shown to be involved in the pathogenesis of diseases including inflammatory bowel disease (IBD), rheumatoid arthritis (RA), Parkinson’s disease (PD), psoriasis, and liver disease through activation of Th17-related pathways (Vernal et al., 2014; Bunte and Beikler, 2019; Feng et al., 2020; Kitamoto et al., 2020; Liu et al., 2021a). It has also been found that recovery of the Th17/Treg balance in periodontitis by the local injection of 3D-exos (a mesenchymal stem cell-derived exosomes, MSC-exos) attenuated experimental colitis. However, further human studies to validate the efficacy and feasibility of Th17-targeted therapies are still lacking.
4 Impact of periodontal bacteria on systemic diseases via the oral-gut axis4.1 Intestinal diseases4.1.1 Inflammatory bowel diseaseIBD is a chronic, recurrent inflammatory disease of the GIT. Periodontitis, also a chronic inflammatory disease, significantly increase the risk of IBD and its disease severity (Vavricka et al., 2013; Lin et al., 2018). Increased periodontal pathogenic bacteria in the gut of IBD patients is positively correlated with the severity of IBD (Lucas López et al., 2017; Schirmer et al., 2018). Animal studies suggest that colitis may be worsened by gut microbial disturbances, which was promoted by gavage of periodontitis salivary microbiota or by infection with periodontal pathogenic bacteria (e.g. P. gingivalis and F. nucleatum) (Cao et al., 2020; Tsuzuno et al., 2021). Successful intestinal colonization of those inoculated oral pathobionts serves as an important trigger to exacerbate IBD. Further study on intestinal colonization, Kitamoto et al. found that a significant level of gut colonization by oral pathobionts was only observed in mice with both oral and gut dysbiosis, rather than only one of the two (Kitamoto and Kamada, 2022b). This implied that periodontal bacteria are pathogenic only to susceptible hosts or promote the progression of pre-existing IBD. Possible pathogenic mechanisms for IBD after intestinal colonization include the ability of periodontal bacteria to compromise the intestinal barrier (downregulation of TJs), enhance intestinal immune responses (e.g. induction of M2 macrophage polarization), and affect intestinal metabolism (e.g. decreased unsaturated fatty acid synthesis and increased arachidonic acid metabolism). Recent evidence suggested that P. gingivalis administration aggravates IBD via a gut microbiota-metabolite linoleic acid (LA)-Th17/Treg cell balance axis. Mechanistically, under Th17-polarizing culture conditions, LA, by specifically binding to AHR as an antagonist, drive Stat1 phosphorylation at Ser727, which in turn represses IL-17 while enhancing Foxp3 expression. Further studies revealed that LA supplementation alleviates P. gingivalis-induced colitis and exacerbation of Th17/Treg cell imbalance (Jia et al., 2024).
4.1.2 Colorectal cancerCRC has the third highest incidence of all cancers (Bray et al., 2018). The gut microbiota of CRC patients is significantly altered compared to that of the healthy population (Chen et al., 2012). Patients with more severe periodontitis have a higher number of oral-derived microorganisms in their gut microbiota and a higher risk of developing CRC, as well as a worse prognosis (Momen-Heravi et al., 2017; Negrut et al., 2023).
In addition to the increased number of oral pathobionts, analysis using 16S rRNA gene sequencing revealed that some of the periodontal bacteria (e.g. F. nucleatum, P. gingivalis, T. denticola, and P. intermedia) were significantly associated with the progression of CRC (Negrut et al., 2023). Animal studies further transplanted the fecal microbiota of periodontitis patients into CRC mice and the same tumor-promoting effect was observed, suggesting that periodontal bacteria may promote CRC by remodelling the oral and gut microbiota (Shi et al., 2023). The current mechanistic study found that in a dysregulated intestinal environment mediated by periodontal bacteria, opportunistic pathogens are able to exploit tumor surface barrier defects, invade normal colonic tissue and induce local inflammation, while producing genotoxic metabolites to induce oncogenic transformation of colonic epithelial cells (Chen et al., 2017).
Many periodontal bacteria are associated with the development of gastrointestinal cancers, with F. nucleatum being most closely associated with CRC (Fan et al., 2018; Brennan and Garrett, 2019). F. nucleatum is usually detected in cancer tissue of CRC patients as well as in secondary distal metastases (e.g. liver and lung) (Flanagan et al., 2014; Abed et al., 2016; Mima et al., 2016; Bullman et al., 2017). Moreover, patients with higher abundance of F. nucleatum in the cancer tissue usually have a shorter survival time and are more likely to recur (Wang et al., 2021). F. nucleatum can be involved in influencing the various stages of CRC development through a variety of mechanisms.
At the stage of tumor progression, F. nucleatum acts to induce a pro-cancer immune microenvironment and assists tumor immune evasion by inhibiting anti-tumor cells as well as increasing the number and function of immunosuppressive cells. For example, F. nucleatum binds and activates the human inhibitory receptors TIGIT and CEACAM1, thereby inhibiting T and NK cells and suppressing anti-tumor immunity (Gur et al., 2015). F. nucleatum also suppresses immunity and increases tumor multiplicity by selectively recruiting tumor-infiltrating myeloid cells (Kostic et al., 2013). In addition to modulating immune cells, F. nucleatum can target tumor cells themselves. F. nucleatum can increase the expression of inflammatory genes (e.g. NF-κB) and oncogenes (e.g. Myc and Cyclin D1) via the FadA/E-cadherin/β-catenin pathway (Rubinstein et al., 2013). This active pathway also leads to F. nucleatum-induced over-expression of chk2, which facilitates DNA damage and tumor growth (Guo et al., 2020). Furthermore, LPS produced by F. nucleatum can upregulate microRNA-21 expression via the TLR4/Myd88/NF-κB pathway, thereby activating the MAPK pathway and enhancing cancer cell proliferation (Yang et al., 2017). In promoting tumor metastasis, F. nucleatum can activate autophagy signaling via the upregulation of CARD3 expression, regulate epithelial-mesenchymal transition (EMT), activate the NF-κB pathway and transmit exosomes (Chen et al., 2020b; Chen S. et al., 2020; Guo et al., 2021; Kong et al., 2021).
During the tumor treatment phase, F. nucleatum increases chemotherapy resistance in CRC patients, ultimately leading to tumor recurrence. F. nucleatum reduces the responsiveness of CRC cells to chemotherapeutic agents (e.g. oxaliplatin and 5-fluorouracil) by upregulating BIRC3 via the TLR4/NF-κB pathway or by inhibiting specific miRNAs involved in autophagy (Yu et al., 2017; Zhang et al., 2019). Therefore, F. nucleatum has been used as a non-invasive biomarker for CRC screening and assessment of prognosis.
Finally, F. nucleatum-based bacteriotherapy may be a potential therapeutic target for CRC. F. nucleatum expresses a variety of virulence factors associated with adhesion (e.g. RadD, Aid1 and Fap2) and invasion (e.g. FadA) of host cells, thereby exerting its pathogenicity through colonization, dissemination, evasion of host defenses and induction of host responses. Some of the F. nucleatum virulence factors have been shown to be closely associated with CRC. For example, FadA promotes the growth of CRC cells, while Fap2 enhances CRC progression by suppressing immune cell activity (Shang and Liu, 2018). An F. nucleatum-specific bacteriophage, FNU1, was found to kill cells and eradicate onco-bacterium from tumor tissue (Wang et al., 2024). In addition, antibiotic inoculation with F. nucleatum could eliminate F. nucleatum from breast cancer and further suppressed F. nucleatum-induced tumor growth (Parhi et al., 2020). Therefore, F. nucleatum (or other oral bacteria)-mediated therapies for CRC may be worth exploring further.
4.2 Rheumatoid arthritisRA is a chronic inflammatory autoimmune disease characterized by bone destruction in multiple joints throughout the body. RA patients often have significant alterations in their oral and gut microbiota that correlate with the severity of joint destruction and the efficacy of antirheumatic treatment in RA patients (Phillips, 2015; Zhang X. et al., 2015; Drago et al., 2019; Möller et al., 2020). Several periodontal bacteria (e.g. P. gingivalis and Prevotella intermedia) are detected in the gut, serum and synovial fluid of RA patients (Martinez-Martinez et al., 2009; Pianta et al., 2017; Drago et al., 2019). RA may be a reactive arthritis aggravated by the repeated translocation of these periodontal bacteria to the joints. Whether oral pathobionts reach the joints through the oral-gut axis or through hematogenous dissemination, most studies suggest that both routes are possible, but more research is needed to determine which route is predominant or prior.
As the most important diagnostic biomarker for RA, serum levels of anti-citrullinated protein antibodies (ACPA) positively correlate with the severity of both RA and periodontitis (Lappin et al., 2013; Bellando-Randone et al., 2021). P. gingivalis can convert arginine residues in proteins to citrulline and increases serum levels of ACPA (Möller et al., 2020). A. actinomycetemcomitans can also indirectly increase protein citrullination through the pathway of leukotoxin (LtxA)-induced peptidyl-arginine deiminase (PAD) dysregulation, thereby increasing ACPA levels (Konig et al., 2016). Thus, one of the important mechanisms by which periodontal bacteria promote RA via the oral-gut axis is chronic exposure to citrullinated protein (CP) triggers the expression of ACPA in joint synovium throughout the body.
In an arthritis mouse model, oral administration of P. gingivalis induced dysbiosis of the gut microbiota, increased IL-6 and CP production in serum, joint and intestinal tissues, and exacerbated joint destruction (Hamamoto et al., 2020). Further, fecal microbiota transplantation (FMT) from P. gingivali-inoculated experimental arthritis mice reproduced donor gut microbiota and resulted in severe joint destruction with increased IL-6 and CP production in joint and intestinal tissues. This suggests that the gut dysbiosis induced by P. gingivalis oral infection may be sufficient to trigger or exacerbate arthritis.
F. nucleatum is also enriched in the gut of RA patients and positively correlate with RA severity (Hong et al., 2023). Mechanistically, F. nucleatum delivers FadA to the joints via outer membrane vesicles (OMVs) and triggers synovial inflammation by activating the Rab5a-YB-1 axis in synovial macrophages. In addition, some studies have treated with the zonulin antagonist larazotide acetate, which specifically increases intestinal barrier integrity (modulating TJs), effectively reduces arthritis onset (Tajik et al., 2020).
The mechanism by which periodontal bacteria promote RA may also be related to the activation of Th17-related pathways. Furthermore, periodontal bacteria (e.g. P. gingivalis and Prevotella nigrescens) are involved in inflammatory bone destruction in RA by increasing the intestinal hyperimmune response triggered by Th17 cellular immune responses, which can induce higher levels of RANKL and TRAP+ osteoclasts (Vernal et al., 2014). P. nigrescens also can reduce the osteoprotective effects of Th2 (de Aquino et al., 2014). All of this evidence demonstrates that the oral-gut axis plays an important role in the influence of periodontal bacteria on RA.
4.3 Brain diseasesPeriodontal bacteria may be indirectly associated with brain diseases through the gut pathway. In addition to ischemic (e.g. stroke) and autoimmune (e.g. multiple sclerosis) brain diseases, evasion strategies of specific pathogens may also alter the function of the blood-brain barrier (BBB) (Daneman and Rescigno, 2009). Here we focus on the pathogenic mechanisms of the oral-gut-brain axis in brain disease, i.e., periodontal bacteria disrupt the gut barrier and BBB, further triggering neuroinflammatory and pathological changes in the brain (Figure 5) (Sansores-España et al., 2021).
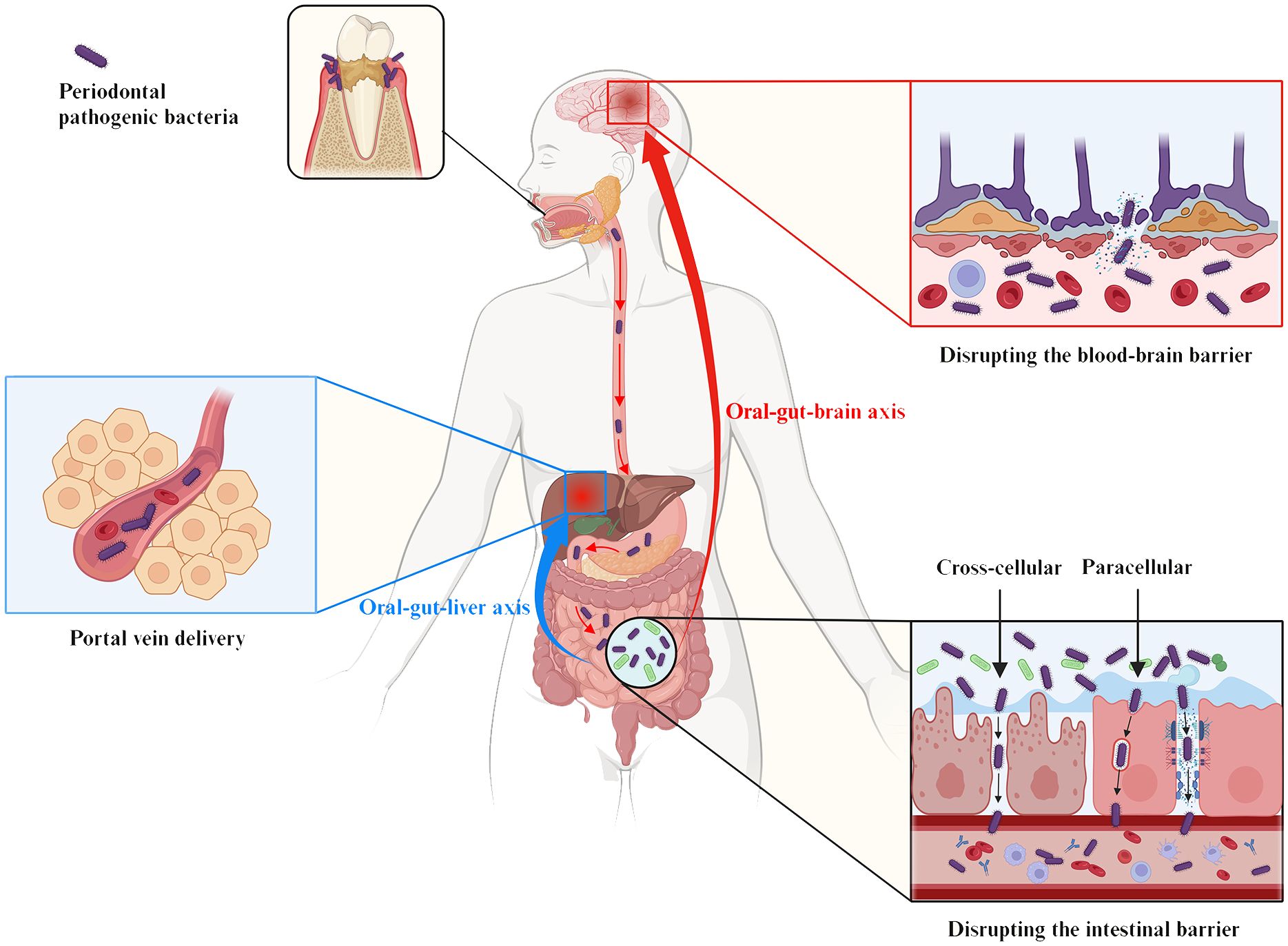
Figure 5. The oral-gut-liver axis and the oral-gut-brain axis serve as the pathophysiologic basis for the influence of periodontal pathogenic bacteria on liver and brain diseases via the intestinal pathway. The gut establishes an anatomical dependence on the liver via portal circulation for the direct delivery of pathogens or metabolites to the liver, a pathophysiologic pathway termed the “oral-gut-hepatic axis” (blue symbols). In addition, the “oral-gut-brain axis” (red symbols) refers to periodontal pathogenic bacteria that trigger neuroinflammatory and pathological changes in the brain based on the disruption of the intestinal barrier and the BBB. As shown, periodontal pathogenic bacteria are involved in the pathogenesis of various systemic diseases by either directly/indirectly reducing intestinal epithelial intercellular junctions (paracellular) or by invading IEC (transcellular), which ultimately affects and breaches the intestinal barrier.
4.3.1 Alzheimer’s diseaseAD is the most common neurodegenerative disease, with progressive behavioral and cognitive impairment as the main clinical feature. Endothelial cells protecting the BBB have been shown to be infected by periodontal bacteria, and virulence factors such as P. gingivalis-LPS and gingipains can even be detected directly in the brains of AD patients (Dominy et al., 2019). Animal studies provide evidence to support this view. Mice with experimental periodontitis showed significant dysbiosis of the gut microbiota, disruption of the intestinal barrier and BBB, and increased levels of LPS in the serum and brain. Meanwhile, neuropathological alterations, including neuronal loss, synaptic injury, and glial activation, as well as progressive cognitive deficits were also observed (Xue et al., 2020).
The pathological mechanisms by which periodontal bacteria promote AD may include the following three aspects. First, periodontal bacteria trigger an excessive accumulation of Aβ in the brain. P. gingivalis may increase Aβ production by releasing gingipains-rich OMVs that drive NLRP3 inflammasome activation, ASC speck aggregation and pyroptotic cell death (Fleetwood et al., 2017; Dominy et al., 2019). Upregulation of advanced glycation end products (RAGE) expression in brain endothelial cells also mediates Aβ influx following P. gingivalis infection (Zeng et al., 2021). In addition, Intestinal-derived LPS, including P. gingivalis-LPS, has been shown to be abundant in the AD brain and has been found to be associated with Aβ plaque formation through activation of the TLR4-mediated NF-κB and MAPK pathways (Zhao et al., 2017).
Second, periodontal bacteria mediate the formation of NFTs in neuronal cells and promote AD (Arnsten et al., 2021). Formation of NFTs results from hyperphosphorylation of the tau protein (Narengaowa et al., 2021). Gingipains can cause proteolysis of tau (tau phosphorylation and cleavage) and subsequent NFTs formation by activating caspase-3. P. gingivalis and its virulence factors, such as lysine-gingipain (Kgp) and arginine-gingipain B (RgpB) are independently and positively correlated with tau load in AD brains (Dominy et al., 2019).
Finally, neuronal necrosis due to neuroinflammation is also a key mechanism for AD. Periodontal bacteria and their virulence factors may activate and accumulate large numbers of microglia through the gut, inducing synaptic toxicity and neuronal death, ultimately exacerbating neurodegeneration (Narengaowa et al., 2021). For example, intraneuronal gingipains may drive neuronal NLRP1 activation, resulting in pyroptosis of neurons and activation of caspase-1, leading to release of the neuroinflammatory interleukins IL-1β and IL-18 (Dominy et al., 2019).
Therapeutically, administration of small-molecule inhibitors of gingipain attenuates neurodegeneration and significantly decreases the host Aβ response to P. gingivalis brain infection, thereby slowing or preventing further accumulation of pathology in AD patients (Dominy et al., 2019). Another study has found a favorable effect of periodontal treatment on AD-related brain atrophy. However, more research is still needed to explore the relationship between periodontal treatment and AD (Schwahn et al., 2022).
4.3.2 Parkinson’s diseaseIntestinal inflammation and leakage promoted by oral administration of periodontal bacteria (e.g. P. gingivalis) may lead to significant loss of dopaminergic neurons and microglial activation in the SNpc, thereby triggering PD (Feng et al., 2020). In animal studies, FMT treatment ameliorated gastrointestinal dysfunction and motor deficits in PD mice by restoring intestinal homeostasis, attenuating damage to the intestinal barrier and BBB and suppressing neuroinflammation and dopaminergic neuronal damage in the substantia nigra (SN) (Zhao Z. et al., 2021). Among the human studies, a recent double-blind, placebo-controlled, randomized trial also suggested that a single FMT induced mild, but long-lasting beneficial effects on motor symptoms in patients with early-stage PD (Bruggeman et al., 2024).
α-synuclein is thought to be a central molecular player involved in the pathogenesis of PD through the oral-gut-brain axis. An endoscopic biopsy study showed a significant correlation between the level of α-synuclein accumulation in neurites of the enteric nervous system and the degree of inflammation of intestinal wall (Stolzenberg et al., 2017). Periodontal bacteria may cross the IEC and induce misfolding and aggregation of α-synuclein in specific enteric neurons. The aggregated α-synuclein will then migrate to the brain via the vagus nerve (Holmqvist et al., 2014; Garrido-Gil et al., 2018). P. gingivalis-mediated activation of leucine-rich repeat kinase 2 (LRRK2) has been shown to consistently induces α-synuclein expression in the gut of R1441G mice, triggering neuroinflammation and subsequent degeneration of dopaminergic neurons (Kozina et al., 2018; Feng et al., 2020).
In addition, IL-17A may also be involved in the effect of periodontal bacteria on PD via the gut (Huang et al., 2014). For example, oral administration of P. gingivalis leads to IL-17A immunoreactivity in the peripheral system and upregulates IL-17RA protein levels in dopaminergic neurons (Feng et al., 2020). Peripheral IL-17A crosses the BBB and mediates dopaminergic neuron degeneration via IL-17RA in microglia (Liu Z. et al., 2019).
4.3.3 Autism spectrum disorderAutism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by abnormal language and interaction skills and repetitive stereotyped behavior (Qiao et al., 2022). Studies have observed disruption of the intestinal barrier and BBB in post-mortem brain tissue and gut in ASD subjects, as well as significant neuroinflammation in the brain (Fiorentino et al., 2016). ASD patients have been further found to have unique oral and gut microbiota distribution patterns, including elevated F/B ratio and increased abundance of opportunistic pathogens, and this bacterial community differences are positively correlated with ASD severity (Tomova et al., 2015; Kong et al., 2019; Johnson et al., 2020). A study that transferred the oral microbiota of ASD donors to an antibiotic-mediated microbiota-depleted mouse model found the induction of ASD-like behaviors (e.g. impaired social behavior) (Kong et al., 2019; Qiao et al., 2022). Significant differences in oral and gut microbiota structure and altered neurosignaling activities, including upregulation of serotonin-related gene expression and TGF-β signaling pathway, were observed in ASD microbiota recipient mice compared to typical development microbiota recipient mice. Furthermore, increased serotonin-related gene expression in ASD microbiota recipient mice was associated with both autistic behaviors and changes in abundance of specific oral microbiota, such as Porphyromonas spp. These evidences highlight the important influence of the oral microbiota in the gut-brain connection.
In addition to these, a variety of other brain disorders, including neuropsychiatric disorders (e.g. depression), neurodegenerative diseases (e.g. multiple sclerosis) and cerebrovascular diseases (e.g. ischemic stroke), may also be associated with the mediating effects of periodontal bacteria via the oral-gut-brain axis pathway (Zhang Z. et al., 2015; Cunha et al., 2019; Maitre et al., 2020; Brown et al., 2021). For example, in a rat model study, oral gavages with P. gingivalis and F. nucleatum simultaneously induced periodontitis, neuroinflammation and depression-like behavior (Martínez et al., 2021). This study further identified F. nucleatum in the frontal cortex of experimental rats and also demonstrated that gastrointestinal translocation of periodontal bacteria can lead to the entry of peripheral LPS into the rat brain and elicit TLR-4-dependent neuroinflammation by finding evidence for the existence of an APOA1-mediated transport mechanism (Martínez et al., 2021). However, clinical studies are still needed for further confirmation.
4.4 Liver diseasesThe gut microbiota may transport bacteria or metabolites directly to the liver via portal circulation (Hou et al., 2022). The impaired gastric acid and bile secretion prevalent in chronic liver disease (e.g. cirrhosis) may also make the gut more susceptible to translocation and colonization by oral bacteria (e.g. P. gingivalis and T. denticola) (Kakiyama et al., 2014; Qin et al., 2014; Wang et al., 2022). The anatomical dependence of the liver on the gut through metabolic exchange and pathogen translocation is therefore the basis for the pathophysiology of periodontal bacteria affecting liver disease via the gut pathway, the “oral-gut-liver axis” referred to in many current studies (Figure 5).
Epidemiological evidence suggests that having severe periodontitis increases the prevalence of non-alcoholic fatty liver disease (NAFLD), which recently renamed Metabolic dysfunction-associated fatty liver disease (MAFLD), mortality associated with cirrhosis, and progression of NAFLD towards fibrotic liver injuries (Akinkugbe et al., 2017; Ladegaard Grønkjær et al., 2018). In addition, patients with hepatocellular carcinoma in combination with periodontitis tend to have worse cancer stage, liver function and prognosis (Tamaki et al., 2011). Pe
留言 (0)