
記住我


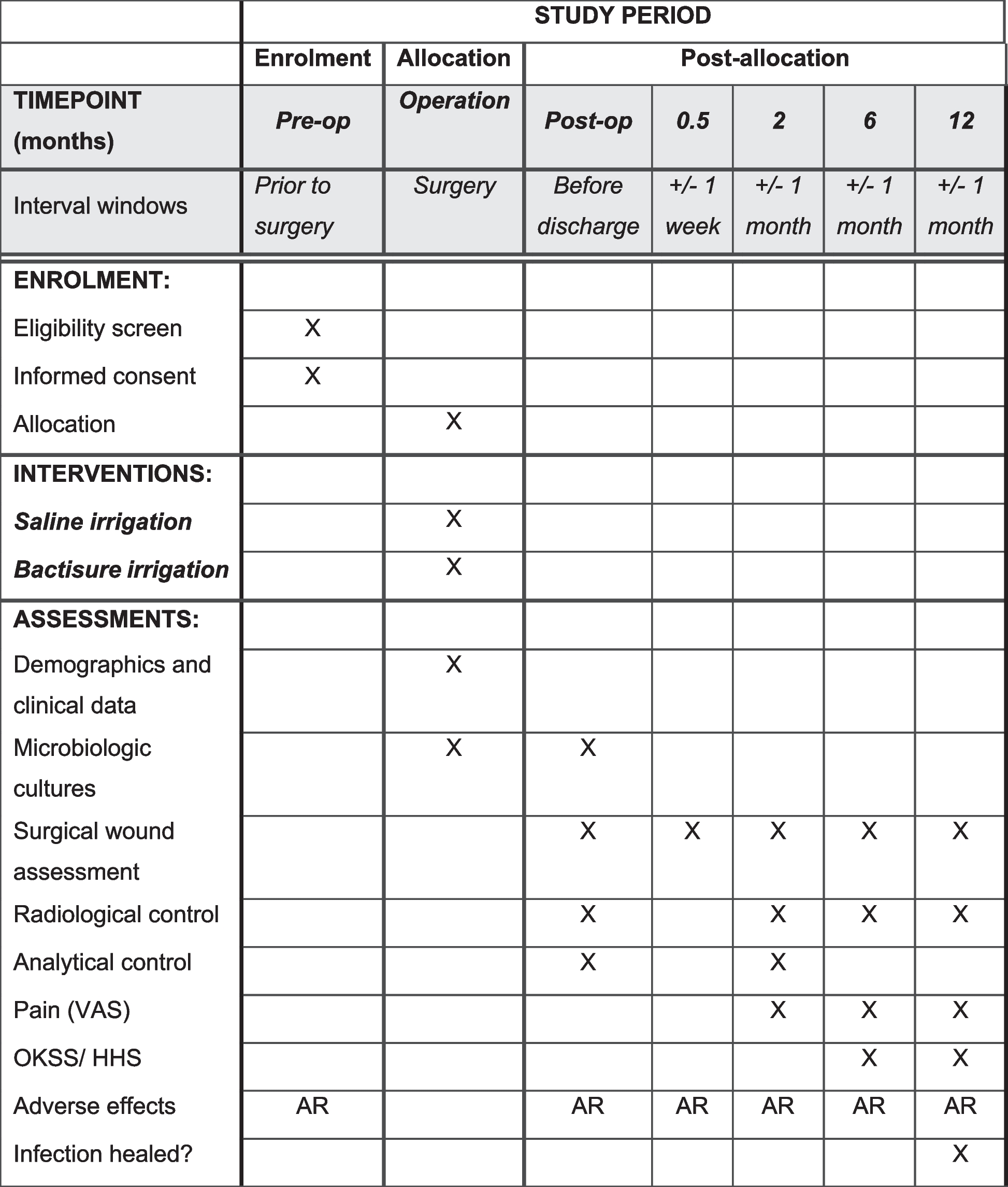
The present study is a phase III parallel randomized double-blind placebo-controlled clinical trial (RCT) among diabetic patients with CKD in the stages before dialysis (stage 3–4). This clinical trial is registered at the Iranian Registry of Clinical Trials (ID: IRCT20170202032367N9). CKD will be confirmed by a nephrologist through the use of laboratory findings. This study is designed to be conducted at Shariati Hospital, affiliated with Tehran University of Medical Sciences, Tehran, Iran. All participants will read and sign the informed consent. We will request consent for review of participants’ medical records and for the collection of blood samples to assess oxidative stress, inflammatory markers, lipid profile, renal function indicators, fasting blood sugar and serum insulin, and serum phosphorous concentration. This study has already been approved by the ethics committee of Tehran University of Medical Sciences (IR.TUMS.SHARIATI.REC.1402.072). We developed the study protocol based on Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 checklist (Supplemental File 1). Timeline of the trial and study flow chart of enrolment, allocation, intervention, and assessment are presented in Table 1 and Figs. 1 and 2, respectively. Before the study is conducted, any changes to the current protocol that have to do with patient safety and welfare, protocol deviations, inadvertent changes that do not impact subject rights, study risk or benefit, data integrity, safety, or welfare must be approved by the Department of Clinical Nutrition and the Medical Ethics Committee of Tehran University of Medical Sciences, Tehran, Iran. All protocol modifications will be reported to the Trials journal (www.trialsjournal.biomedcentral.com). The significant number of exclusion criteria should be underlined as a reason for the researchers to stay longer at Shariati Hospital to recruit enough participants and achieve the desired sample size.
Table 1 Timeline of the trialFig. 1 Fig. 2
Fig. 2 Inclusion criteria
Inclusion criteriaIn this study, we will recruit diabetic patients with CKD in the stages before dialysis (stage 3-4) which is confirmed by a nephrologist through the use of laboratory findings; aged 25-65 years with a body mass index (BMI) > 20 and < 30 kg/m2; and who have willingness to cooperate in the study.
Non-inclusion criteriaPatients will not be included if they (1) have autoimmune kidney disease or glomerulonephritis; (2) have blood pressure (BP) > 160/100 mmHg; (3) are pregnant or lactating women or have a plan to get pregnant in the next 6 months; (4) have infectious, inflammatory diseases, thyroid gland disorders, and thrombocytopenia; (5) are under enteral and parenteral nutritional support; (6) have a history of taking omega-3 and antioxidant supplements (vitamin E, vitamin C, vitamin B6, selenium, zinc, and beta-carotene separately) from 3 months before entering the study; (7) are taking glucocorticoids with a dose of more than 5 mg, antibiotics, fluvoxamine, non-steroidal anti-inflammatory drugs (NSAIDs), and warfarin; (8) are smokers; and (9) have night shift jobs.
Exclusion criteriaWe will exclude the following patients: (1) those who do not want to continue taking supplements, (2) those who get pregnant throughout the study; (3) those who did not consume more than 10% of supplements in each follow-up; and (4) those who enter the stage of dialysis or kidney transplantation.
Study designIndividuals who meet the inclusion criteria will be enrolled. After recruitment, participants will pass a 2-week run-in period, during which three 24-h food recalls (2 working days and 1 weekend day) and two 1-day physical activity records will be completed for each patient to collect demographic and dietary information. To measure biochemical parameters, 10-mL overnight fasting venous blood samples will be obtained from each patient. At the end of run-in period, anthropometric measures, oxidative stress and inflammatory biomarkers, lipid profile, glycemic status, renal function, blood pressure, serum phosphorous level, and sleep quality will be evaluated.
RandomizationAfter recruiting participants, we will use stratified block randomization, in which participants will be stratified based on age (under 30 and above 30) and sex (male/female) into different blocks. For each patient in a certain block, a matched person in terms of age and sex would be placed in other block. Then, the two patients in a single block would be randomly assigned into the intervention and control groups. We will employ a 1:1 allocation ratio to ensure equal representation in both groups. Furthermore, our study is designed as a superiority trial. A third person who is not aware of the study’s aim will randomly allocate participants using a computer-generated random sequence (sequentially numbered).
BlindingAll patients, researchers, nephrologist, and laboratory staff will all be blind to the intervention. The supplements used in both groups are identical in shape, color, and smell. The supplements will be given to both groups packing in similar containers. The supplement cans separated by the letters A or B. The blinding code will be unknown until the end of the study except in emergency cases. Study participants will take their bottles in two times: at their first visit and in the middle of the trial, at week 5.
Study interventionPatients in the experimental group (N = 24) will receive 5 mg melatonin supplements twice a day for 10 weeks. Patients in the control group (N = 24) will consume placebo tablets containing starch. Participants in both groups will be requested to take the tablets before bedtime for 10 weeks. Weight, size, shape, taste, color, smell, and lot number are the same in melatonin and placebo capsules. Even the patients in the placebo group will not deprived of their main medical treatment during the study. There is no anticipated harm and compensation for trial participation; if any of the participants have a problem, necessary treatments will be done for them. Safety of melatonin supplementation was confirmed by study pharmacist. All subjects will be asked not to change their routine daily diet, physical activity, and medicines. There will be no special criteria for discontinuing or modifying allocated interventions. Melatonin or placebo will not require alteration to usual care pathways (including use of any medication) and these will continue for both trial arms. If any of the participants does not consume 10% of their capsules, this state is considered as a research dropout and is excluded from the study.
OutcomesPrimary and secondary outcomesThe main primary outcome in the current study would be oxidative stress biomarkers including total antioxidant capacity (TAC), total oxidative stress (TOS), and malondialdehyde (MDA) as well as inflammatory markers including interleukin-6 (IL-6) and highly sensitive C reactive protein (hs-CRP). The study secondary outcome variables would be lipid profile (total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL), high-density lipoprotein (HDL)), renal function indicators (blood urea nitrogen (BUN), creatinine, uric acid), fasting blood sugar and serum insulin, systolic and diastolic blood pressure (SBP and DBP), serum phosphorous concentration, sleep quality, weight, BMI, WC, energy intake, and consumption of energy-contributing nutrients including carbohydrates, fat, and protein.
Measurements and assessmentsDietary intake and physical activityTo assess dietary intakes throughout the study, they will be asked to fill three 1-day dietary recalls at the beginning of the study, weeks 5 and 10 of intervention. Dietary recall format is presented in the online supplemental file. We will compute food and nutrient intake of study participants based on the examination of these dietary recalls. The Nutritionist 4 software, which was modified for Iranian foods, will be used to perform this calculation.
In addition, two 1-day physical activity records will be gathered from all participants to examine the difference in activity between two groups. In order to analyze physical activity records, we will use MET-h/day values for each type of physical activity, based on published guidelines [20], considering the time spent by each participant.
Anthropometric measuresData on anthropometric indices including body weight, height, WC, and BMI will be collected at study baseline and end of the trial. Participants will be weighed in a fasting state, without shoes with minimal clothing to the nearest 0.1 kg accuracy, using a digital scale. Standing height will be measured using a standard stadiometer without shoes with an accuracy of 0.5 cm. WC will be measured to the nearest 0.1 cm accuracy by non-stretching tape measure around the abdomen at the distance between the suprailiac bone and the last rib. BMI will be calculated by the measured height and weight (weight (kg)/height (m2)).
Clinical outcomesSystolic and diastolic blood pressures will be measured twice at the right arm using a mercury barometer calibrated by the Institute of Standardization and Industrial Research, with a 15-min interval in between measurements, while the patient is sitting quietly for 5 min. The average of the two measurements will be analyzed to calculate the systolic and diastolic blood pressures. Before starting the intervention and at the end of trial, the sleep quality of the patients will be evaluated using a self-rated questionnaire which assesses sleep quality and disturbances over a 1-month time interval. Nineteen individual items generate seven “component” scores. The sum of scores for these seven components yields one global score: Pittsburgh Sleep Quality Index (PSQI) [21].
Other biochemical variablesAt the study baseline and after the intervention, a 10-mL venous blood sample will be taken from each person after 12-h fasting. Then, serum will be isolated from whole blood by centrifugation for 10 min at 3500 rpm and for further analysis serum samples will be stored at − 80 °C. The process of accessing serum concentrations of inflammatory factors and oxidative stress biomarkers (TAC, TOS, MDA, IL-6, hs-CRP) will be done by the enzyme-linked immunosorbent assay (ELISA) commercial kits.
Sample size calculationConsidering the type I error of 5% (α = 0.05) and type II error of 20% (β = 0.20, power = 80%) and total antioxidant capacity (TAC) as the key variable [13], we manually, without the use of any software, calculated required sample size using the following formula:

n = sample size in each group
α = type 1 error = 0.05
β = power = 80%
S 12 = variance of the intervention group = (173)2
S 22 = variance of the control group = (108)2
∆2 = minimal clinically important difference = (86)2
Based on this formula, we reached a sample size of 22 people in each group. Considering a 10% likelihood of dropping out, the number of participants in each group increased to 24 people.
Data management and monitoringA clinical trial monitor occasionally supervises the study progress, ensures patient rights and well-being are safeguarded, the protocol, ethical requirements, standards, and regulations are being followed, the essential documentation is available, and collected data are accurate as there were recorded. One of the investigators will check the coding, security, and storage of data. In addition, he/she will evaluate data entry and data values twice. If any participant reports the occurrence of adverse events, more information is required to make decision about excluding the participants from the trial. Unblinding is permissible in this situation based on the Medical Ethics Committee criteria.
Adherence to the interventionThe study progress will be pursued by calling the patients once a week to ensure that they regularly consume the tablets. Adherence to the intervention will be checked by counting the returned tablets at the half and end of the trial visits. Compliance rate will be computed according to the following formula and poor compliance will be considered as less than 90% [22].
$$\mathrm\;\mathrm:\;\left(\mathrm\;\mathrm/\mathrm\;\mathrm\right)\times100$$
Statistical analysisStatistical analysis will be conducted using the SPSS software (version 25, SPSS Inc., Chicago, IL, USA). The Kolmogorov–Smirnov test will be applied to examine the normality of data. Moreover, we will use chi-square test and Fisher’s exact test to compare categorical variables. Also, independent sample t-test and Wilcoxon rank-sum test will be applied to compare continuous variables within-group, whereas we will use paired sample t-test and Mann–Whitney U test for between group comparisons. Normally distributed variables will be reported as the mean and standard deviation, while the median and interquartile range (IQR) will be used for reporting non-normally distributed variables. To compare the differences in primary and secondary outcomes between the two study groups at the end of the trial and also adjust the final findings for potential confounders, we will apply the analysis of covariance (ANCOVA) test. Subgroup analysis or adjusted analysis will not be applicable. A p value less than 0.05 will be regarded as statistically significant. If we have missing data, statistical analysis will be done using the intention to treat (ITT) method by imputation.
留言 (0)