Cell culture
The mouse brain endothelial (bEnd.3) (American Type Culture Collection (ATCC®) CRL2299™) and human glioblastoma (U87) (ATCC® HTB-14 ™) cell lines were used in the current study. The bEnd.3 cell line was used as the BBB model, while the conditioned media was extracted from the U87 cell line. Both cell lines were cultured under aseptic conditions in Dulbecco’s Modified Eagles Medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% foetal calf serum (FCS) (Sigma-Aldrich), and 1% of penicillin–streptomycin (Thermo Fisher Scientific) to create a complete culture medium. Cells were grown in a 95% humidified incubator (HF 212 UV incubator (Heal Force®, Shanghai China)) at 37 °C and 5% CO2.
Glioblastoma-conditioned media extraction and preparation
To prepare the CM, U87 cells were cultured and maintained in a 75 cm2 cell culture flask in the above-mentioned conditions until 80–90% confluence. Thereafter, the cells were detached using TrypLE™ Express Enzyme (Thermo Fisher Scientific) and seeded into 75 cm2 culture flasks alongside 15 mL of complete media at a concentration of 2 × 104 cells/mL for 48 h (h), after which CM was collected (referred to as U87-CM hereafter). The collected U87-CM was centrifuged using the Beckman Allegra X-14/R Series Benchtop Centrifuge (Beckman Coulter, Brea, USA) at 1,000 × g for 10 min to remove cells and cellular debris. Thereafter the U87-CM was filtered through a 0.2 μm sterile syringe filter and stored at − 20 °C. Before treatment, the U87-CM was thawed at room temperature (21 °C) and diluted to 40% with complete media to replenish nutrients.
Brain endothelial cell growth following glioblastoma cell and CM exposure
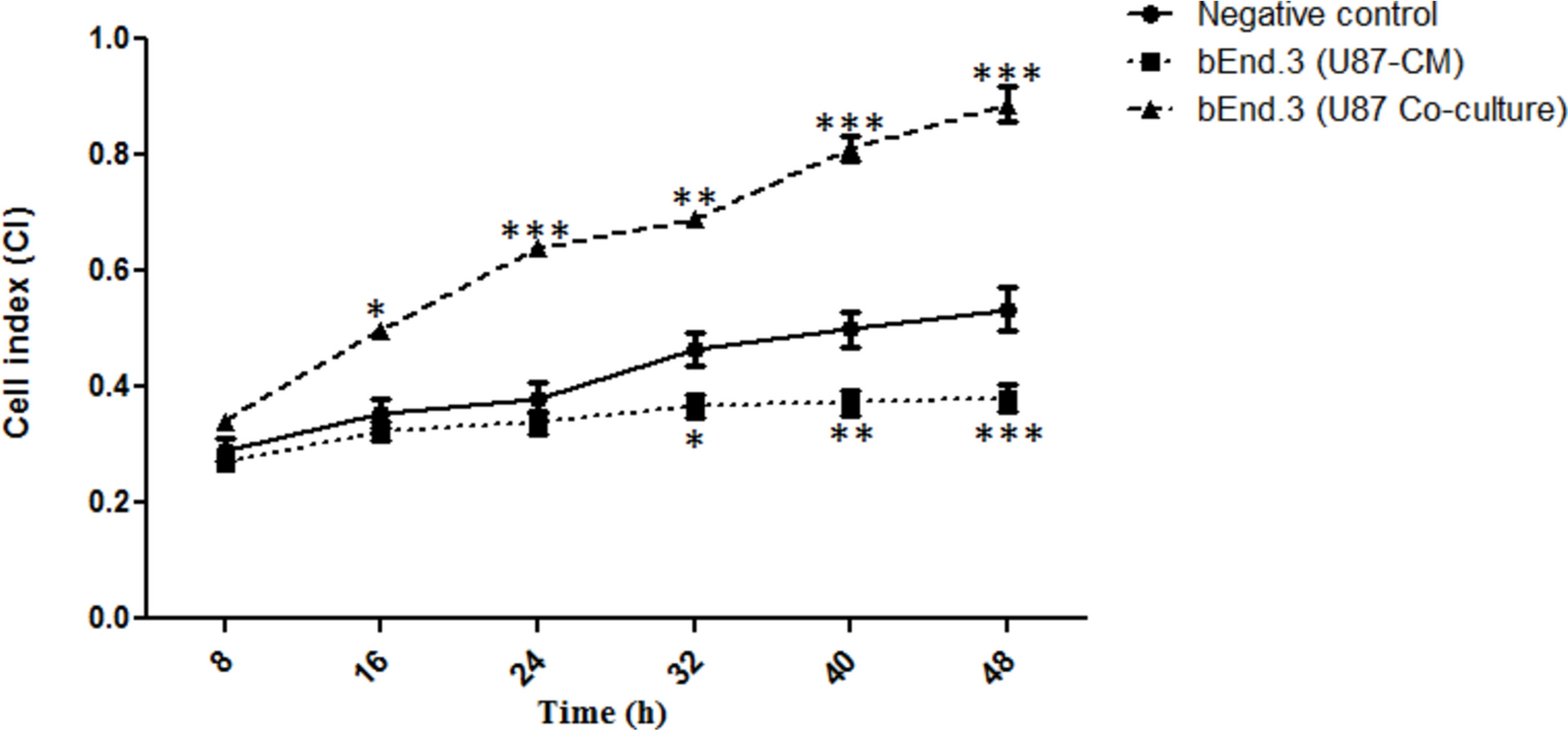
The growth of bEnd.3 cells treated either with U87-CM or co-cultured with U87 cells was assessed using the xCELLigence Real-Time Cell Analysis Dual Plate (RTCA-DP) system (Acea Biosciences, CA, USA). The xCELLigence assay was adapted from the method described by Mabeta and Pepper (2015) [16]. Two different experimental setups were assessed: i) a mono-culture of bEnd.3 cells treated with U87-CM, and ii) a co-culture of bEnd.3 and U87 cells. The experimental setup included a negative control of an untreated mono-culture of bEnd.3 cells. For the mono-cultures, 50 μL of complete culture medium was added to all wells of the 16-well E-plate to blank the plates. The E-plates were then inserted into the xCELLigence RTCA (ACEA BioSciences), and background measurements were recorded for the wells before adding 50 μL of a bEnd.3 cell suspension at 2 × 104 cells/mL into each well. The background measurement allows the software to automatically report any connection problems or plate defects.
The bEnd.3 cells were then allowed to attach for 24 h, before treatment with 50 μL of U87-CM. For the co-culture, the same blanking process was conducted using complete culture media. Following the blanking process, 2 × 104 cells/mL bEnd.3 cell suspension was added to the bottom chamber of each well to a total volume of 100 μL. Following 24 h of attachment, inserts were assembled on top of the bottom chambers, and 60 μL of U87 cell suspension was added at 2 × 104 cells/mL into each well. The cells were allowed to settle for 30 min inside the laminar flow at room temperature before the E-plates were re-engaged onto the xCELLigence RTCA plate station (Acea Biosciences, CA, USA) and incubated for 48 h at 37 °C and 5% CO2. The cell index (CI) values were recorded continuously for 48 h. In the context of this study, CI is defined as the changes in cell-electrode impedance. The xCELLigence system measures the changes in impedance and converts them into CI.
Migration of brain endothelial cells exposed to glioblastoma CM
The migration of bEnd.3 cells treated with U87-CM was assessed according to a previously described method from Mabeta and Pepper (2009) [17]. In the current study, bEnd.3 cells were seeded at a concentration of 2 × 104 cells/mL in 1 mL of complete media in each well of a 12-well plate. Once the bEnd.3 cells reached confluence, a straight cross-sectional line scratch wound was gently made in the centre of the plate using a 200 μL pipette tip to detach central cells. The plate was rinsed twice with sterile phosphate-buffered saline (PBS) to remove the floating cells and debris. Cells were then treated with a U87-CM diluted with serum-free media, while the negative control cells were treated with serum-free media. Due to the experimental setup of the scratch migration assay, it was not conducted for the co-culture model. In addition, the cells would have been disturbed during imaging, as it requires prior removal of inserts and reassembling. The plates were then incubated in a 95% humidified incubator at 37 °C and 5% CO2. Images were then captured at 0, 24, and 48 h using a Zeiss Axiovert CFL40 phase-contrast microscope (Carl Zeiss AG, Oberkochen, Germany) at 5X magnification with the Zeiss Axiovert MRm monochrome camera. The width of each scratch was measured using the ImageJ software 1.53 K (National Institutes of Health and the Laboratory for Optical and Computational Instrumentation (LOCI, University of Wisconsin) Wisconsin, USA). The following formula was used to calculate the percentage of wound closure:
$$} }\% = \frac} = 0} \right) - \left( } = \Delta }} \right)} \right)}}} = 0} \right)}} } 100$$
where At = 0 is the initial wound area, At = Δt, is the wound area after n hours of the initial scratch.
Permeability of brain endothelial cells exposed to U87 cells or CM
The validation of junctional tightness was assessed through the measurement of the paracellular compound sodium fluorescein (NaFl) according to a modified method from Czupalla and colleagues (2014) [18]. The NaFl molecule has a small size and molecular weight (376 Da) amenable to permeability studies, and has been validated for such purposes [19]. Two models were evaluated for permeability in the current study: i) bEnd.3 mono-culture treated with U87-CM, and ii) bEnd.3 cells co-cultured with U87 cells. The permeability of bEnd.3 cell monolayer was determined using a transwell permeability assay. The transwell permeability assay quantifies the movement of a fluorescent tracer over a cell monolayer.
The bEnd.3 cells were seeded at a density of 5 × 104 cells/mL into Thincert™ inserts (volume: 150 μL) and the U87 cells were seeded at a density of 2 × 104 cells/mL in 1 mL of complete media into a separate 24-well plate. Plates were allowed to attach for 24 h in a 95% humidified incubator at 37 °C and 5% CO2. Following 24 h inserts were transferred to their respective model setups: i) a 24-well plate with U87-CM, and ii) inserts transferred onto 24-well plate containing U87 cells seeded at the bottom of the wells. The plates were incubated for 48 h. After 48 h the inserts were transferred to a 24-transwell plate containing 600 μL of fresh complete culture medium.
A 200 μL aliquot of 0.1 mg/mL NaFl solution was added to the upper chamber of the inserts. Every 15 min for 60 min, 100 μL of media was removed from wells and transferred into the wells of a black 96-well plate. Fluorescence from the media was read at 480/560 nm (excitation/emission wavelength) using the Synergy II plate reader (BioTek Inc.). The fluorescent marker endothelial apparent permeability (Papp) was calculated as follows:
$$} = \frac} } (RFU)}}} } (\min ) } } } (}^ ) } } } } } (})}}$$
Tight junction protein expression in brain endothelial cells exposed to U87 cells or CM
The bEnd.3 cells were seeded at a density of 5 × 104 cells/mL in 2 mL of complete media in a 6-well plate and incubated in a 95% humidified incubator at 37 °C and 5% CO2 for 24 h for attachment. Following 24 h of attachment, the bEnd.3 cells were treated with U87-CM and incubated further for 48 and 72 h. In addition, bEnd.3 cells were also seeded at a density of 5 × 104 cells/mL in 1 mL media into a 24-well plate, while a density of 2 × 104 cells/mL U87 cells was seeded into inserts placed in a separate 24-well plate and were allowed to attach for 24 h. Following 24 h of attachment, the inserts containing U87 were transferred and placed onto the bEnd.3 containing 24-well plate and were incubated further for 48 and 72 h.
Following 48 and 72 h, bEnd.3 cells were washed with PBS and trypsinised using TrypLE™ Express. The trypsinised cells were collected in a 15 mL tube and centrifuged at 200 × g for 5 min. Following centrifugation cells were counted using a haemocytometer. Thereafter, cells in the 15 mL tube were re-suspended in ice-cold PBS and centrifuged again at 200 × g for 5 min. The supernatant was discarded and the cell pellet was re-suspended with 1 mL ice-cold (per 1 × 107 cells/mL) radioimmunoprecipitation assay (RIPA) buffer with HALT protease inhibitor, and gently agitated on ice for 30 min on the shaker. The lysed cells were transferred into a micro-centrifuge tube and centrifuged using the Beckman Allegra X-14/R Series Benchtop Centrifuge (Beckman Coulter, Brea, USA) at 16,000 × g for 20 min at 4 °C. Following centrifugation, the supernatant was aspirated and the pellet was discarded. An aliquot of 25 µL of cell lysate was collected and protein concentration was determined using the bicinchoninic acid protein determination assay according to a modified method from Smith and colleagues (1985) [20]. The cell lysates were then aliquoted and stored at − 204 °C.
The TJ protein expression was then determined using a Western blot assay. Cell lysates were thawed at room temperature, and a 20 µg sample was equally mixed with sodium dodecyl sulphate (SDS) sample buffer (2 × Laemmli buffer) and incubated for 5 min at 954 °C. The resultant sample was centrifuged at 16,000 × g for 1 min. Equal amounts of the protein (20 µg) were loaded into the wells and separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) for 5 min at 60 V. The voltage was then increased to 120 V and ran for 1 h. Following the run, the gel was transferred using a transfer buffer onto a polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was rinsed and stained with Ponceau S stain to assess the quality of the transfer. The PVDF membrane blot was blocked with 3% BSA in Tris-buffered saline with tween 20 (TBST) buffer and incubated for 1 h at room temperature.
The expressions of occludin, claudin-5, and ZO-1 were detected using mouse monoclonal primary antibodies against occludin (mouse anti-occludin (clone: OC-3F10), Thermo Fisher Scientific) (1:500), claudin-5 (mouse anti-claudin-5 (clone: 4C3C2), Thermo Fisher Scientific) (1:500), ZO-1 (mouse anti-ZO-1 (clone: ZO1-1A12), Thermo Fisher Scientific) (1:500), included in the blocking buffer incubated overnight at 44 °C. β-actin was used as the housekeeping protein to account for errors in loading or protein transfer using anti-β-actin antibody (mouse anti-β-actin (clone: AC-15), Sigma-Aldrich) (1:100,000). Thereafter, the membrane was incubated with goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, horseradish peroxidase (goat anti-mouse IgG) (Thermo Fisher Scientific) (1:10,000) in a blocking buffer for 1 h at room temperature. The blot membranes were washed three times with TBST before imaging. The immunoreactive bands were detected by the chemiluminescence using the Clarity™ Western ECL Substrates (Bio-Rad), through the ChemiDocMP (Bio-Rad, California, USA) imager and analysed with Image Lab 6.0 software (Bio-Rad, California, USA).
Statistical analysis
All experiments were conducted with at least three biological and three technical replicates, resulting in nine data sets per experimental condition. The raw data was captured using Microsoft Excel software and analysed using the two-way repeated-measures analysis of variance (ANOVA) with the post-hoc Bonferroni and Holm-Sidak test through GraphPad Prism 8.0 software. The results were presented as mean ± standard deviation (SD). The statistical significance was set at p ≤ 0.05 compared to the means of the negative control.



留言 (0)