
記住我
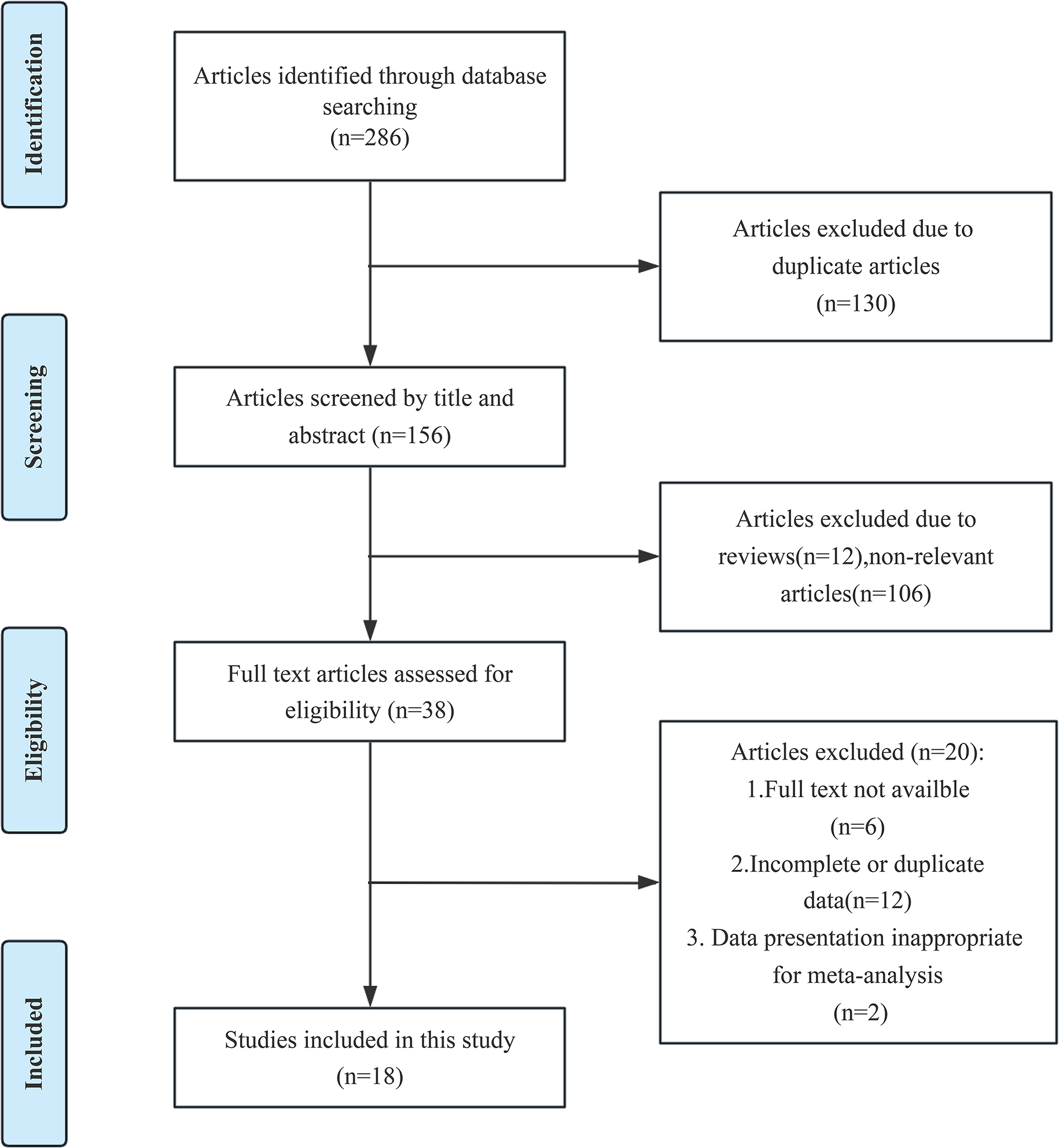
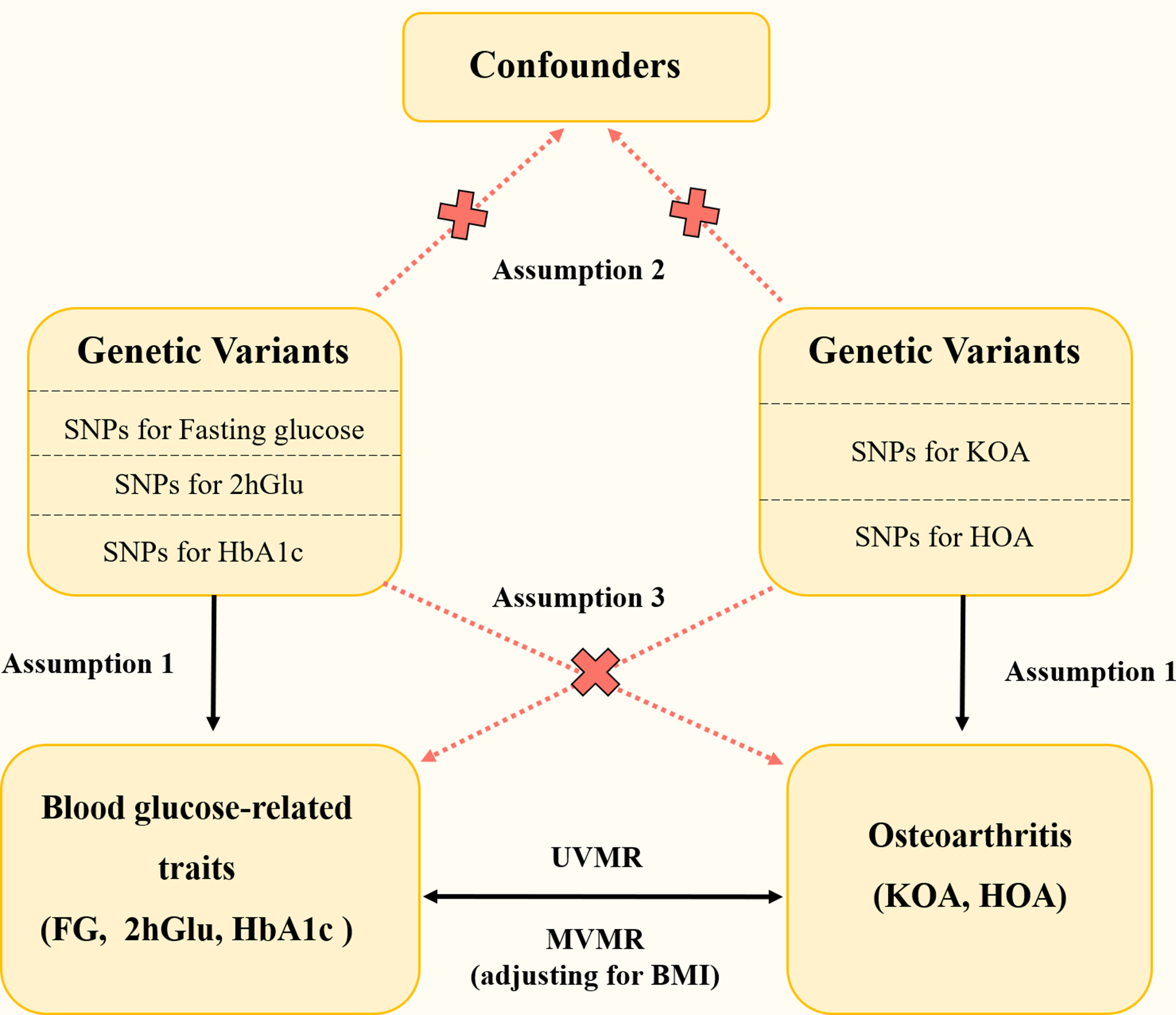

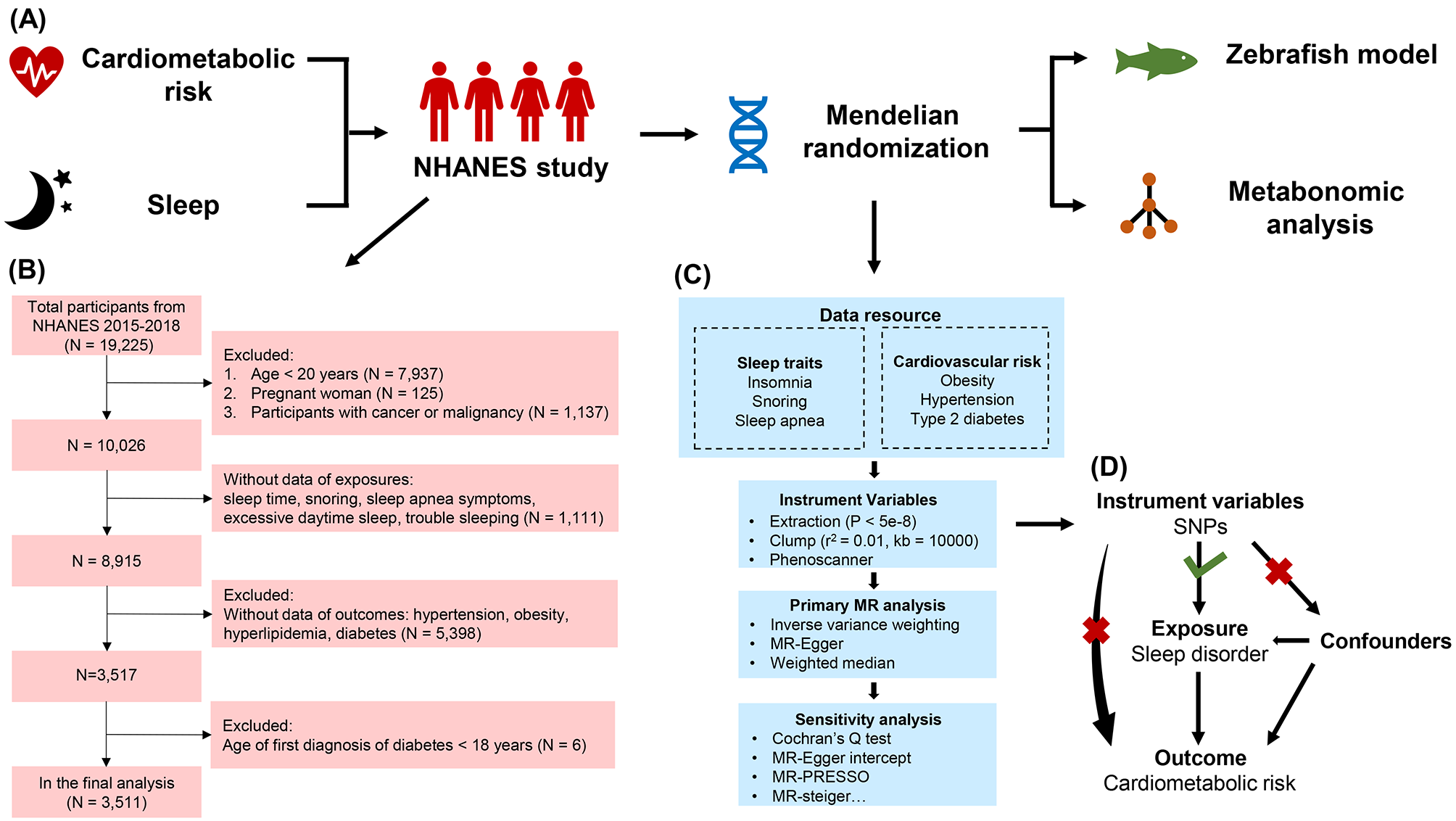
As shown in Fig. 1A, this study first explored the relationship between sleep problems and cardiometabolic risks through the NHANES database. MR analysis was conducted to explore the causal relationship between sleep problems and cardiometabolic risks based on the significant results of cross-sectional NHANES studies. To further explore the underlying mechanism bridging the association of sleep problems and cardiometabolic risks, zebrafish models of sleep disorders were constructed and sent for metabolomics analysis to discover altered metabolic pathways.
Fig. 1
Research scheme and flowchart of data process. (A) The schematic illustration comprised the NHANES study, Mendelian randomization analysis, zebrafish model construction, and metabolomics analysis. (B) The flowchart of the participants from NHANES 2015–2018. (C) Data resource and study design of our MR analysis. (D) Three assumptions in the MR analysis
NHANES analysisNHANES is an epidemiological survey project that utilizes multistage, complex, and probabilistic sampling methods conducted by the National Health Survey Center (NCHS). It aims to assess the health and nutritional status of adults and children in the United States.
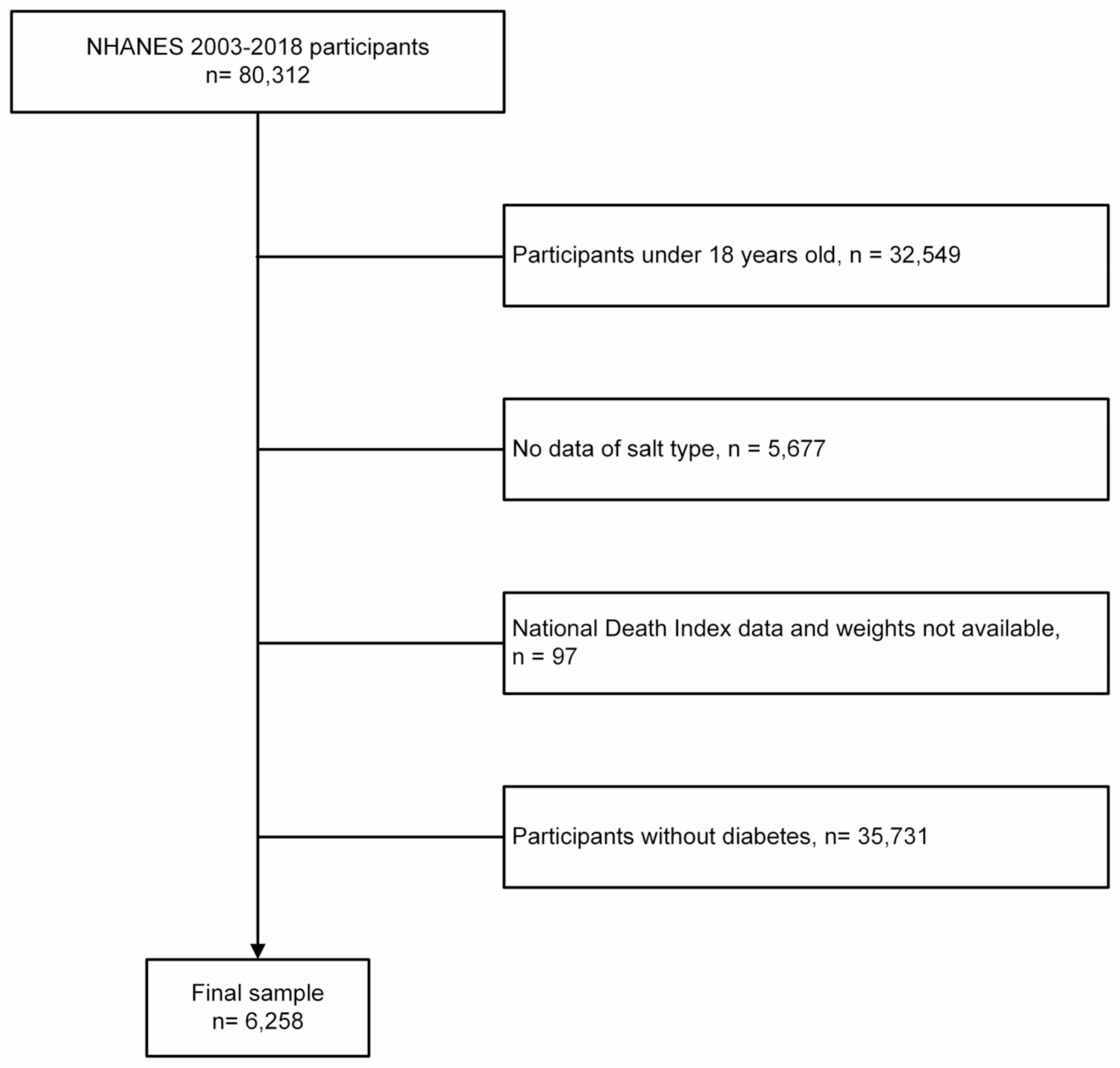
The data used in this study was from NHANES 2015–2018. As presented in Fig. 1B, the participants aged < 20 years or pregnant or with cancer or malignancy were excluded.
Participants with missing data on exposures and outcomes were subsequently excluded, as were participants < 18 years old at the time of their first diagnosis of diabetes.
NHANES has obtained permission from the Institutional Review Committee of the Centers for Disease Control and Prevention (CDC), and all participants have signed informed consent forms before the investigation.
Definition of exposures, outcomes, and covariablesThe five sleep behaviors (sleep time, snoring, sleep apnea symptoms, excessive daytime sleep, and trouble sleeping) obtained from self-report in the questionnaire were defined as the exposure variables. The outcomes were defined as cardiometabolic risk factors, which included hypertension, obesity, hyperlipidemia, and T2DM.
Covariates included age, marital status, sex (biological sex, male/female), education level, race, triglyceride (TG), physical activity category, total calories taken, alcohol intake, family income-to-poverty ratio (PIR), sedentary time, and tobacco exposure.
Statistical analysisAll analyses used weighted sampling weights, stratification, and primary sampling units to make the results more nationally representative. The standard for selecting covariates was to change the effect value by more than 10%. The missing variables of the covariate were divided into a separate group and named “Missing”. Data were described as means and SDs for normally distributed continuous variables and as medians and interquartile ranges for nonnormally distributed continuous variables. The chi-square test was used for categorical variables (%). Three weighted logistic regression models (model 1: adjusted no variables; model 2: adjusted age, race, sex; model 3: adjusted sex, race, age, education level, marital status, TG, physical activity category, total calories taken, alcohol intake, PIR, sedentary time, and tobacco exposure) were employed to calculate the regression odds ratio (OR) and 95% confidence intervals (CIs) between five sleep problems and cardiometabolic risk factors.
All data analyses were conducted using software R (version 4.2.1, R core team) and EmpowerStats (version 4.0, EmpowerStats core team). P < 0.05 (two-sided) was considered significant.
MR analysisAs shown in Fig. 1C, a bi-directional MR study was conducted, following the Strengthening the Reporting of Observational Studies in Epidemiology reporting guideline for MR (STROBE-MR), to explore the potential causal relationship between three sleep problems (insomnia, snoring, sleep apnea) and three cardiovascular risks (obesity, hypertension, T2DM), which has been preliminarily identified in the NHANES study. Single-nucleotide polymorphism (SNP) was extracted as instrument variables (IVs) from previous summary-level GWAS data on sleep apnea [13], snoring [14], and insomnia [14]. While GWAS data on snoring and insomnia was generated from the UK biobank, GWAS data on sleep apnea was from a meta-analysis across five independent cohorts from the United Kingdom (UKB), Canada (Canadian Longitudinal Study of Aging, CLSA), the United States (Partners Healthcare Biobank), Australia (Australian Genetics of Depression Study, AGDS), and Finland (FinnGen) [13]. GWAS data on insomnia, snoring, and sleep apnea included the following covariates, namely, age, sex, and genetic principal components. GWAS summary statistics of outcomes were acquired from the research of the FinnGen consortium [15]. The eligibility criteria, methods of assessment, diagnostic criteria for diseases, ethics committee approval, and participants’ consent can be found in the original articles. We checked the sample description of each GWAS study to ensure that the GWAS data used for exposure was independent of those for the outcomes. For snoring and insomnia, all participants were from the UK biobank, indicating there wasn’t a sample overlap between snoring-cardiometabolic risks or insomnia-cardiometabolic risks. For sleep apnea, 66,216 participants were from the FinnGen consortium, leading to a sample overlap proportion of 12.65% (66216/523366). For IVs to be valid, selected IVs should conform to three assumptions in Fig. 1D: (1) SNPs were robustly associated with sleep problems; (2) SNPs were not related to confounders; (3) SNPs did not affect the cardiometabolic risks except through the potential effects of the sleep problems. After harmonization, a bi-directional MR was performed to calculate the MR estimate of each sleep problem-cardiovascular risk pair by multiple methods, including Inverse weighted median (IVW), Weighted median (WME), and MR-Egger. Comprehensive sensitivity analyses such as heterogeneity, pleiotropy, and directionality were conducted to exclude potential bias.
Instrumental variables selectionSNPs closely (P < 5 × 10− 8) associated with sleep problems were extracted as candidate IVs. After SNPs were extracted at a significance level of P < 5 × 10− 8, they were clumped (r2 < 0.01, kb > 10000, population = EUR) to guarantee the independence of each SNP. SNPs that directly related to outcomes were identified by Phenoscanner and were removed to reduce pleiotropy bias. Palindromic SNPs were subsequently excluded, and the F statistic of each IV was calculated. Sample sizes, number of controls and cases, population, and consortium of the GWAS were listed in Table 1.
Table 1 Characteristics of included GWAS summary-level data of sleep, cardiovascular risksMendelian randomization studyAfter the harmonization, a two-sample MR analysis by the Two-Sample MR R package (version 0.5.6) was performed. The IVW method was used to calculate combined association across the Wald ratios for all IVs. Although the IVW method possessed strong statistical power, this method required all included instruments to be valid. Therefore, we also included results of MR-Egger, WME, and weighted mode providing unbiased reference even in the presence of invalid IVs [16]. Results of IVW with multiplicative random effect were referred to in the presence of heterogeneity. The mean F-statistics of each exposure generated from the calculation of the F statistic of each SNP was applied to exclude potential weak-instrument bias. Moreover, the significance P value was set as 0.0167 (0.05/3) based on bonferroni correction to exclude multiple-test bias.
The MR-PRESSO test by MR-PRESSO R package (1.0) was conducted to exclude horizontal pleiotropic outliers for the pleiotropy correction. The MR-Egger intercept result was considered an additional reference of pleiotropy. MR-PRESSO results after excluding outliers were referred to in the presence of pleiotropy. The Q tests of IVW and MR-Egger were performed to identify potential heterogeneity. In addition, the directionality between the exposure and outcome was validated by the results of the MR-Steiger analysis. Leave-one-analysis recalculated the MR estimate after excluding SNPs individually to identify whether a single SNP drove the bias of MR results. All data analyses were conducted using software R (version 4.2.1, R core team). The procedures of reverse MR analysis were the same.
Experimental animals-zebrafishZebrafish husbandry and construction of sleep disorder modelAdult wild-type AB zebrafish were cultured in a circulating water system at a temperature of 28.5 ± 1℃. They were fed three times daily and given a 14:10 h light-dark (LD) cycle. Pair male and female zebrafish on the night before and lay eggs within 1 h of turning on the light the following day to obtain embryos. The obtained embryos were placed in a light-controlled (14:10 h LD) incubator at 28.5℃, and cultured with Holt Buffer containing 0.5 mg/L methylene blue.
Embryos that were well developed at 1-day postfertilization (dpf) were selected and divided into two groups. The control group was given a 14:10 h LD cycle lasting three days. The experimental group was given a 24 h light-light (LL) environment for three days to construct a sleep disorder model.
Analysis of behavior after building the sleep-disorder modelAfter the construction of the model, 144 (repeat three times with 48 larvae each group) larvae in each group were randomly selected for motor behavior detection. The movement of zebrafish was tracked using the zebrafish behavior analysis system, and two environments of 14:10 h LD and 24 h dark-dark (DD) were set, respectively.
Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)Samples (repeat three times with 30 larvae each group) every 4 h for a total of 24 h were collected, and the total RNA content was determined by NanoDrop (NanoDrop, DE, USA). The purity and quality of RNA were determined by 260/280nm ratio and agarose gel electrophoresis. The first-strand cDNA was synthesized by the Prime-Script RT Reagent Kit. Furthermore, relevant instruments (StepOnePlus™ Real-Time PCR System, ABI) were used for RT–qPCRs. Internal reference group β-actin, with the 2−ΔΔCT method, was applied to calculate the relative changes of each gene (bmal1b, cry1aa, cry1ab, clock1a, per1b, per2). The primers for each gene are shown in Supplementary Table S1.
Widely targeted metabolomics analysisThe 150 larvae (repeat three times with 50 larvae each group) from the control group (normal light-dark cycle, 14:10 h LD) and experimental group (continuous light cycle for 3 days, 24 h LL) were collected at 9:00 and 21:00, respectively, for targeted metabolomics analysis. Use R software to perform principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and orthogonal partial least squares discriminant analysis (OPLS-DA) dimensionality reduction analysis on the samples. Metabolites with P value < 0.05 and VIP value > 1 were selected (considered to have significant statistical differences).
Statistical analysisSPSS 26.0 and GraphPad Prism 8.4 were used for statistical analysis. If the P value of the variance homogeneity test is greater than 0.05, the T-test is used. If the P value of the result of the homogeneity test of variance is less than 0.05, the T2 test is used. P < 0.05 was deemed statistically significant.
留言 (0)