
記住我
Climate change poses an increasing risk to productivity, welfare, and requires farm management practices change without compromising effectiveness in the livestock industry (Rhoads et al., 2009; Yang et al., 2021). Moreover, economically important traits are significantly influenced by genotype-by-environment interactions, and the animals’ performance can deteriorate with rising global temperatures (Haile-Mariam et al., 2008; Braz et al., 2021). Over the past few decades, intensive breeding for greater productivity, such as increased milk yield, growth rate, little size, and body weight in livestock, led to higher metabolic heat production (Cabezón et al., 2017), potentially reducing the ability of livestock (i.e., pigs) to thrive in harsh environments. Consequently, this has caused farm management to become more challenging with declining profitability as animals face heightened risks from the severe effects of global warming. Thus, enhancing CR has become a primary objective in livestock breeding.
Climate resilience refers to the animal’s ability to maintain or quickly return to euthermia under thermally stressful conditions (Colditz and Hine, 2016; Wen et al., 2024). Many studies have investigated CR and proposed novel indicators, such as genetic variance of the slope of reaction norm models (Shi et al., 2021; Waters et al., 2022; Freitas et al., 2024). However, most direct resilience phenotypes are difficult or expensive to measure, leading to less frequent measurements and more issues, such as low phenotypic variability and low to moderate heritability estimates (Guy et al., 2012; Gorssen et al., 2021). In previous studies, various phenotypes related to heat stress (HS) in lactating sows—including vaginal temperature, respiration rate, skin surface temperature, hair density, and body condition score—were measured under HS conditions and considered as useful indicators of HS (Scheffer et al., 2018; Gorssen et al., 2021; Johnson et al., 2023). These phenotypes exhibited low to moderate heritability estimates. However, identifying CR of animals based on these measures alone is challenging. Individuals who exhibit greater consistency in their phenotypes over time are likely to have higher resilience (Scheffer et al., 2018; Berghof et al., 2019). This is because they are expected to deviate less from their optimal production or physiological levels when faced with disruptions, leading to increased survival and reduced disease incidence (Scheffer et al., 2018; Berghof et al., 2019).
Increased availability of longitudinal data from various methods, such as automatic thermometers, feeding stations, and computer vision systems, makes it possible to derive more effective resilience indicators (Chen et al., 2023; Pedrosa et al., 2023). For instance, methods for deriving several new resilience indicators in dairy cattle based on deviations from observed and expected performance, including variance, lag-1 autocorrelation, and skewness of deviations, have been proposed (Poppe et al., 2020). These methods have been applied in resilience studies across various species, including cattle (Poppe et al., 2020; Chen et al., 2023), pigs (Mancin et al., 2024), and dairy goats (Sánchez-Molano et al., 2019).
Fifteen novel CR indicators, such as variance, lag-1 autocorrelation, and skewness of deviations, as well as HS duration, using longitudinal automatically-recorded vaginal temperature were developed in our previous study (Wen et al., 2024). Most of these indicators were moderately heritable and had low to high genetic correlations with each other. Current understanding of the biological mechanisms and genetic factors influencing CR in lactating sows is rather limited. In this context, genome-wide association studies (GWAS) enable the detection of single nucleotide polymorphisms (SNP) associated with traits of interest (Visscher et al., 2012). Many GWAS studies focusing on resilience have been conducted in different species, such as chicken (Doekes et al., 2023), pigs (Putz et al., 2019), sheep (Tsartsianidou et al., 2021), and cattle (Alonso-Hearn et al., 2022; Chen et al., 2024). However, CR is expected to be a polygenic trait influenced by numerous biological mechanisms, which could lead to the identification of many putative quantitative trait loci (QTL), some of them with small effect and located on different chromosomes. GWAS can contribute to a better understanding of the genetic basis underlying phenotypic variability in CR. By undertaking GWAS on different CR metrics, we can delve deeper into the genetic basis of this complex trait, potentially uncovering valuable insights that will not only advance our scientific knowledge but also inform breeding strategies aimed at enhancing CR in sows. Thus, the primary study objectives were to 1) detect SNPs and genomic regions significantly associated with twelve CR indicators derived from automatically-recorded vaginal temperature measured in lactating sows under HS conditions; and 2) identify the underlying biological functions and metabolic pathways these regions are involved in based on functional genomic analyses.
2 Materials and methods2.1 DatasetsAll live animal data collection procedures were approved by the Purdue University Animal Care and Use Committee (Protocol #1912001990). All data collection procedures, physiological data, genotype information, and quality control processes have been previously described in our previous studies (Johnson et al., 2023; Wen et al., 2023; Wen et al., 2024). In brief, 1,639 lactating sows (parities 2–7; Landrace × Large White) were genotyped using the PorcineSNP50K Bead Chip (Illumina, San Diego, CA, United States). The vaginal temperature (TV) of 1,381 sows within the studied population was automatically measured every 10 min from June 5th to July 30th, 2021, using a vaginally implanted thermochron data recorder (Johnson et al., 2023). Ambient temperature and humidity of each barn was automatically collected every 5 minutes (Johnson et al., 2023). The phenotypic and genomic quality control procedures performed can be accessed on our research (Johnson et al., 2023; Wen et al., 2023). Twelve CR indicators were derived based on variability in automatically measured vaginal temperature (Wen et al., 2024). Log-transformed variance [LnVar(Ave) and LnVar(Med)], Lag-1 autocorrelation (Autocor (Ave) and Autocor(Med)], and skewness [Skew(Ave) and Skew(Med)] of the deviations between observed and the average (Ave) or median (Med) values from moving windows consisting of six consecutive observations with a 10-minute interval were calculated for each animal (Poppe et al., 2020). The HS thresholds for individuals under distinct ventilation conditions (mechanical ventilation at 39.76°C and natural ventilation at 39.78°C) were reported (Johnson et al., 2023). Additional traits were derived including the daily maximum vaginal temperature (MaxTv) per individual and the HS duration (HSD), which quantifies the duration of the time interval in which an individual’s vaginal temperature consistently exceeded the HS threshold each day. Two CR indicators were normalized median (Nor_medvar) or average TV (Nor_avevar) multiplied by the normalized TV variance on a population level as follows,
Nor_medvari=Medi−MedminMedmax−Medmin×varTvi−varTvminvarTvmax−varTvmin and Nor_avevari=Avei−AveminAvemax−Avemin×varTvi−varTvminvarTvmax−varTvmin, where Medi, Avei, and VarTvi represent the median, average, and variance of TV for individual i, Avemin and Avemax are the minimum and maximum median TV, Medmin and Medmax are the minimum and maximum median TV, and VarTvmin and VarTvmax are the minimum and maximum TV variance, respectively. Furthermore, two additional traits were derived based on the total deviations between TV and HS threshold values, which were calculated by summing up the TV values above (HSUA) or below (HSUB) the HS threshold throughout the entire data collection period as follows,
HSUA=∑t=1nTvt−HS threshold and HSUB=∑t=1nTvt−HS threshold, where Tvt is the TV at time point t. All these CR indicators were described in detail (Wen et al., 2024), and the heritability estimates ranged from 0.084 ± 0.037 [Skew (Med)] to 0.291 ± 0.047 (HSUB).
2.2 Genome-wide association studies and functional genomic analysesGenome-wide association studies between the CR indicators and the SNPs were conducted using the linear mixed animal model in the GCTA software (Yang et al., 2011), with the option of leaving one chromosome out (MLMA-LOCO). The effects included in the GWAS models are the same as those reported previously (Wen et al., 2024). After performing the GWAS, the genomic inflation factor (λ) was calculated to evaluate potential bias in the results, e.g., from unaccounted population stratification. The λ value was calculated as the ratio of the median of the observed distribution of the statistic to the expected median, for which a 95% confidence interval of value was further derived (Devlin and Roeder, 1999). The Bonferroni correction was used for multiple testing corrections (Armstrong, 2014). The genome-wide significance and suggestive significance threshold were set as 1.17×10−6 (P = 0.05/N) and 2.34×10−5 (P = 1/N), respectively, where N represents the total SNP number left after removing SNPs based on linkage disequilibrium (LD) (indep-pairwise 50 5 0.1, N = 42,729). To avoid type I errors and false negative results, another less-stringent significance threshold of 0.05 divided by the number of independent chromosomal segments (Me) at chromosome-wise levels was considered (Li et al., 2015), following the model: Me=2NeLlog NeL, where Me is an function of effective population size (Ne) and chromosome length (L, in centimorgans–cM). Ne was considered to be equal to 60 (Hall, 2016) and 1 cM equivalent to 1 Mb (Wang et al., 2016). Quantile-quantile plots (Q-Q plots) were created using the CMplot R package (Yin et al., 2021).
The GALLO R package (Fonseca et al., 2020) was used to detect genes located within 500 Kb up and downstream of significant SNP and QTL regions previously cited in the pig QTLdb (Hu et al., 2019) based on the latest genome reference Sscrofa 11.1 assembly (http://useast.ensembl.org/Sus_scrofa/Info/Index). Gene Ontology (GO) (Ashburner et al., 2000) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) enrichment analyses for candidate genes were carried out using the DAVID platform (Huang et al., 2009).
3 Results and discussion3.1 GWAS results summaryWe first conducted GWAS studies for all traits to investigate the genetic basis and biological mechanisms associated with heritable CR indicators [heritability estimates ranging from 0.084 ± 0.037 to 0.291 ± 0.047 (Wen et al., 2024)]. The traits evaluated were LnVar(Ave), Autocor(Ave), Skew(Ave), LnVar(Med), Autocor(Med), Skew(Med), MaxTv, HSD, Nor_avevar, Nor_medvar, HSUA, and HSUB. The genomic inflation factors ranged from 0.95 to 1.2 for all indicators, showing small inflation of P-values for the estimated SNP effects (Price et al., 2010). Lambda values and Q-Q plots for each CR trait are shown in Figure 1.
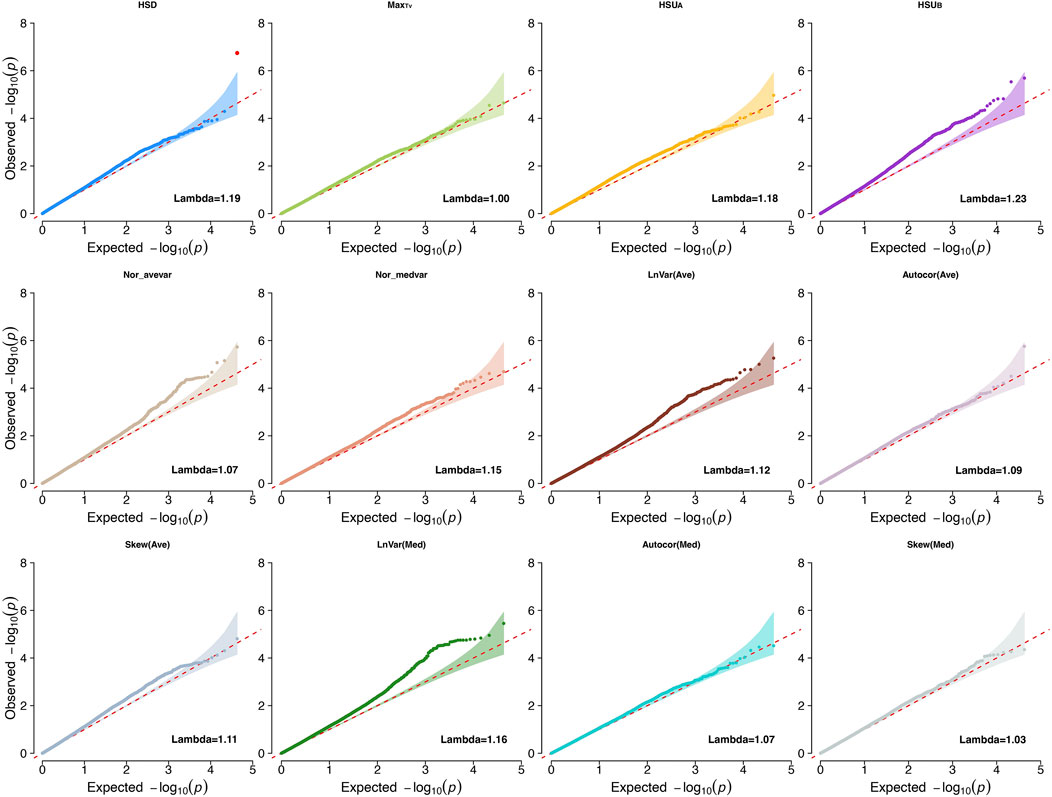
Figure 1. Quantile-quantile plots (QQ-plot) and lambda values for the climatic resilience indicators evaluated1. 1Indicators: LnVar(Ave), log-transformed variance of the deviations between each observation and the average values from moving windows that contains six continuous observations with 10-mins interval in between; Autocor (Ave): Lag-1 autocorrelation of the deviations between the average values from moving windows that contains six continuous observations with 10-mins interval in between; Skew (Ave): skewness of the deviations between each observation and the average values from moving windows that contains six continuous observations with 10-mins interval in between; LnVar(Med): log-transformed variance of the deviations between the median values from moving windows that contains six continuous observations with 10-mins interval in between; Autocor (Med): Lag-1 autocorrelation of the deviations between the median values from moving windows that contains six continuous observations with 10-mins interval in between; Skew (Med): skewness of the deviations between each observation and the median values from moving windows that contains six continuous observations with 10-mins interval in between; Nor_avevar: normalized average TV multiplies the normalized TV variance; Nor_medvar: normalized median TV multiplies the normalized TV variance; HSUA: sum of TV values above the HS threshold during the whole data collection period; HSUB: sum of TV values below the HS threshold during the whole data collection period; HSD: The length of time during which the body temperature remained above the HS threshold value for each collection day; MaxTv: The highest TV of each measurement day.
Thirty-one SNPs located on nine Sus scrofa chromosomes (SSC) that reached at least the suggestive significance level were detected for nine CR indicators and presented in Table 1. Four, one, one, 13, one, four, one, five, one SNP at the suggestive threshold was detected for LnVar(Ave), Autocor (Ave), Skew (Ave), LnVar (Med), Nor_medvar, Nor_avevar, HSUA, HSUB, and HSD, respectively. Among these SNPs, one SNP located on SSC15:135,366,143 bp, one SNP on SSC6:16,449,770 bp, one SNP on SSC6:16,323,291 bp, and three SNPs on SSC2:88,327,932 bp and 88,631,882 bp and SSC3:18,088,863 bp, were identified as significant at the chromosome-wise level threshold for four CR indicators: Autocor (Ave), LnVar(Med), HSUB, and Nor_avevar, respectively. Notably, only one SNP (SSC9: 15,692,376 bp) for HSD met the most stringent significance thresholds (Table 1). No significant associations were found for MaxTv, Autocor (Med), and Skew (Med), and this may be lack of power. The small number of suggestive SNPs for each indicator in our study indicates that the evaluated CR indicators are highly polygenic, with many genomic regions of small effects located throughout the different chromosomes. Most HS or heat tolerance related traits in livestock are polygenic (Macciotta et al., 2017; Tiezzi et al., 2020; Cheruiyot et al., 2021), with few major genes identified. Larger sample sizes could be beneficial for identifying these QTLs with smaller effects. These findings are in agreement with our previous research (Freitas et al., 2023; Wen et al., 2023).
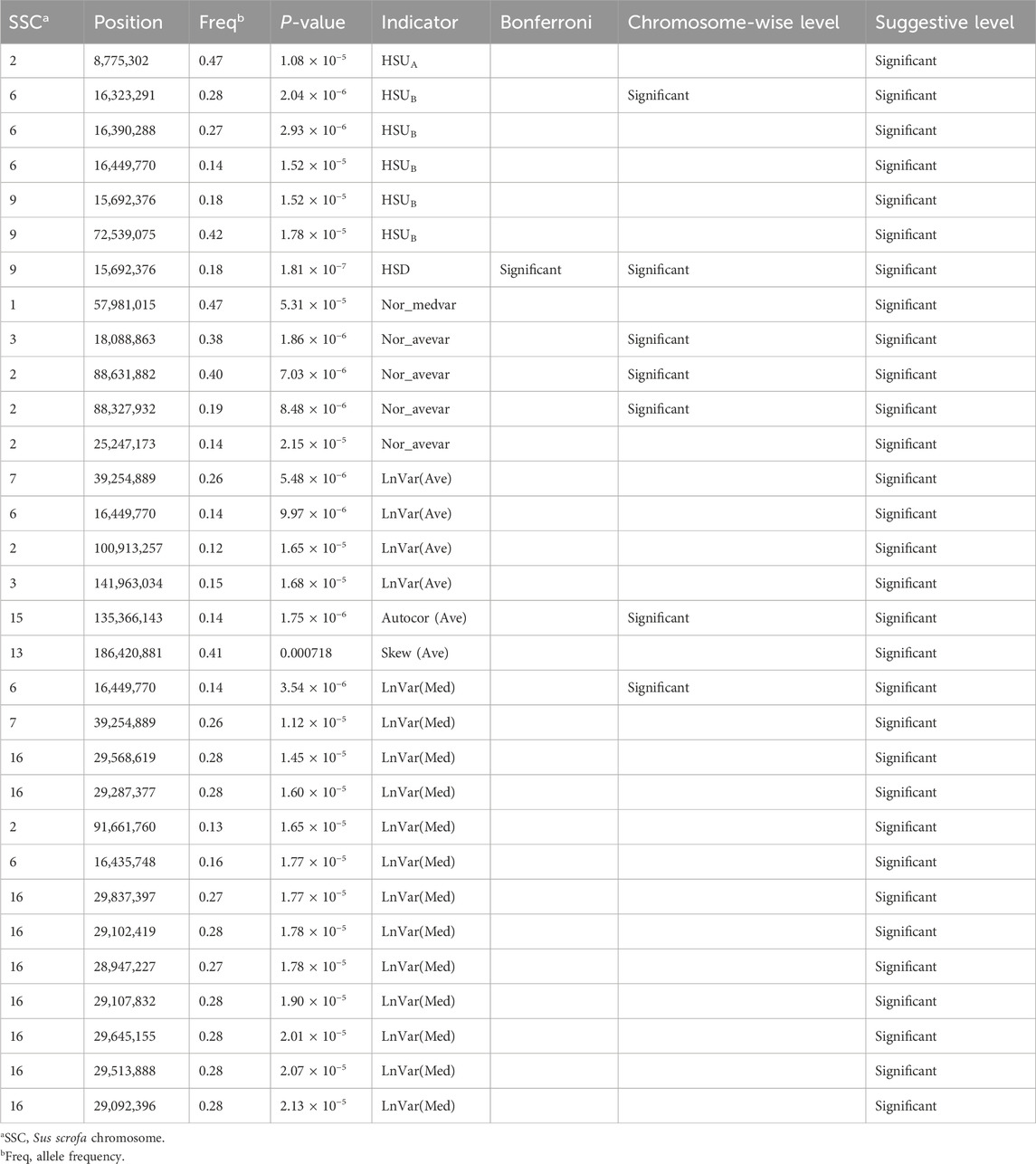
Table 1. Candidate SNPs that reached the suggestive significance level for CR indicators.
Four common SNPs were identified to be associated with more than one CR indicator. The SNPs are located on SSC6:16,449,770 bp and SSC6:16,435,748 bp [HSUB, LnVar(Ave), and LnVar(Med)], SSC7:39,254,889 bp [LnVar(Ave) and LnVar(Med)], and SSC9:15,692,376 (HSUB and HSD). The CR indicators created based on similar metrics are highly correlated at the genetic level, such as LnVar(Ave) and LnVar(Med), or HSUA and HSUB (Wen et al., 2024). Interestingly, the SNPs identified for these correlated indicators are not distributed at similar genomic regions. There are several reasons for that: first, as previously mentioned, these indicators are highly polygenic, and we did not find any major gene control in the CR indicators; Second, the candidate SNPs identified by each indicator are in linkage disequilibrium with their causal variants, explaining why overlapping SNPs are still observed across different indicators.
3.2 Candidate genes and functional genomic analysesA total of 442 positional genes harboring or adjacent to the significant SNPs were mapped, including 212 protein-coding genes, 225 non-coding RNAs, and 5 pseudogenes (Supplementary Additional File S1: Supplementary Table S1). Specifically, 22, three, one, 30, one, 91, 45, 18, and one protein-coding genes were identified to be associated with LnVar(Ave), Autocor(Ave), Skew(Ave), LnVar(Med), Nor_medvar, Nor_avevar, HSUA, HSUB, and HSD, respectively. For LnVar(Ave), the genomic regions around four significant SNPs (SSC2: 100,913,257 bp; SSC3: 141,963,034 bp; SSC6: 16,449,770 bp; SSC7: 39,254,889 bp) harbors Heat Shock Protein 90 Alpha Family Class B Member 1 (HSP90AB1), Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel 1 (HCN1), SPT3 Homolog SAGA And STAGA Complex Component (SUPT3H), and Transmembrane Protein 63B (TMEM63B) genes. HSP90AB1 functions as a chaperone and plays a role in protein transport and degradation. Its expression level decreased significantly in response to HS in pigs (Seibert et al., 2019). Besides, one SNP (SNP g.4338T > C) within HSP90AB1 was found to be significantly related to heat tolerance in Thai indigenous cattle (Charoensook et al., 2012). HCN1, SUPT3H, and TMEM63B were found to be associated with cellular and oxidative stress response in salmon (Beemelmanns et al., 2021), milk production in Holstein cattle (Liu et al., 2021), and residual feed intake in purebred French Large White pigs (Messad et al., 2021), respectively.
One SNP, located at 135,366,143 bp on SSC15, was associated with Autocor (Ave) at chromosome-wise and suggestive significance level. The genomic region around this SNP contains SH3 Domain Binding Protein 4 (SH3BP4) and ArfGAP with GTPase Domain, Ankyrin Repeat and PH Domain 1 (AGAP1). SH3BP4 was identified in a region with copy number variation in South African Nguni cattle, which are recognized for their ability to sustain harsh environmental conditions and resistance to parasites and disease (Wang et al., 2015). There were a few peaks with significant SNPs for LnVar(Med) on SSC2: 91,661,760 bp, SSC6: 16.435–16.449 Mb, SSC7: 39,254,889 bp, and between 28.947 and 29.837 Mb on SSC16. The up and downstream of the significant SNPs covered 15 candidate genes that were enriched for LnVar(Ave) before due to the overlapping SNPs. Strong associations were found on SSC2 and SSC3, with Low-Density Lipoprotein Receptor Class A Domain Containing 3 (LDLRAD3), solute carrier family 1 member 2 (SLC1A2), Dimethylglycine Dehydrogenase (DMGDH), Betaine-Homocysteine S-Methyltransferase 2 (BHMT2), and Homer Scaffold Protein 1 (HOMER1), harboring the most significant SNPs for Nor_avevar. LDLRAD3, known to encode a low-density lipoprotein (LDL) receptor and associated with decreased levels of very low-density lipoprotein receptor, has been linked to HS in chickens (Jastrebski et al., 2017; Wang et al., 2020). Further research in mice has revealed that the very low-density lipoprotein receptor plays a crucial role in regulating thermogenesis in brown adipocytes, suggesting its importance in body temperature regulation (Shin et al., 2022). It has been demonstrated that genes encoding the very low-density lipoprotein receptor are crucial for both lipid metabolism and the response to temperature stress (Álvarez et al., 2020). SLC1A2 was significantly downregulated in the mouse pituitary gland under hot conditions and was related to stress response (Memon et al., 2016). DMGDH was considered a candidate gene for heat tolerance, defined as the rate of decline (slope) in milk, fat, and protein yield in swamp buffalos. Furthermore, DMGDH may be involved in alleviating oxidative stress in heat-stressed cattle (Cheruiyot et al., 2021). BHMT2 is involved in regulating homocysteine metabolism with beneficial effects in heat-stressed animals through its activity against osmotic stress and protection of protein denaturation (Cottrell et al., 2015; Del Vesco et al., 2015). Besides, BHMT2 has been identified as a positively selected candidate gene affecting thermotolerance in African indigenous cattle (Ankole, Ogaden, N'Dama, Boran, and Kenana cattle), using XP-CLR and XP-EHH population statistics (Taye et al., 2017). HOMER1 plays an important role in behavior, particularly concerning adaptation to stress and fear responses (Kamprath et al., 2009). For Skew(Ave) and Nor_medvar, only one protein-coding gene was identified for each trait–ENSSSCG00000042482 and ENSSSCG00000052428, respectively. However, no information regarding their functions was found for these two genes.
The genes Solute Carrier Family 3 Member 2 (SLC3A2), Syntaxin 5 (STX5), RNA Polymerase II Subunit G (POLR2G), and Glucosidase II Alpha Subunit (GANAB) were significantly enriched for HSUA. Previous research observed downregulated SLC3A2 gene expression in bovine mammary epithelial cells under HS conditions (Ma et al., 2018), and this might be an adaptive response to meet increased amino acid requirements during HS (Rhoads et al., 2011). A frameshift mutation in STX5 has been considered a potential causal mutation for cattle’s heat tolerance, and it also significantly impacts milk production (Cheruiyot et al., 2021). Besides, STX5 was linked to tick resistance in Belmont Red cattle (Tabor et al., 2017) and Tunisian indigenous sheep (Ahbara et al., 2022). Tick burdens might correlate with thermal comfort (Rocha et al., 2019), as traits such as skin thickness, hair density, and skin secretions influence both tick resistance and heat regulation (Shyma et al., 2015). GANAB was found to be downregulated in jejunum mucosa of German Holstein cows under HS conditions, and this is related to responses to incorrect protein folding and stabilization processes (Koch et al., 2021). For HSUB, Cytochrome P450 Family 51 Subfamily A Member 1 (CYP51A1), and Cyclin Dependent Kinase 6 (CDK6) were detected in the SSC9. CYP51A1, a gene involved in cholesterol and sterol metabolism, was observed to be upregulated in the plasma of laying hens in response to HS (Zhu et al., 2019). CDK6 was significantly downregulated by HS in duck granulosa cells (Yang et al., 2021).
Only one protein-coding gene (ENSSSCG00000034240) was annotated for the only significant SNP of HSD. The limited overlap between candidate genes identified for various CR indicators in this study and candidate genes from GWAS of resilience for HS is not surprising. First, the traits we used to define resilience, such as HSD and LnVar(Med), differ from those in many other studies (Cheruiyot et al., 2021; Tsartsianidou et al., 2021). Additionally, automatically measured TV enables us to get more accurate CR indicator values. Given the complexity of CR that spans a broad spectrum of adaptive responses, from behavioral to physiological to cellular, it is likely that varying QTLs are captured based on the indicators employed in GWAS studies.
To investigate the biological functions of these candidate genes further, we performed GO and KEGG analysis using DAVID, as shown in Tables 2, 3. Two, one, and one significant KEGG pathways were observed for Lnvar(Med), Lnvar(Ave), and Nor_avevar, respectively. These pathways are related to stress response [e.g., chemical carcinogenesis - receptor activation (Trush and Kensler, 1991)], immune and inflammatory responses [e.g., Th17 cell differentiation (Guo et al., 2018)], cell survival, proliferation, and migration [e.g., PI3K/Akt signaling pathway (Zhang et al., 2016; Yiming et al., 2021)], and nervous system (e.g., Glutamatergic synapse) (Niciu et al., 2012). Heat stress also has been documented to cause a change in animals’ adaptive immune function, transitioning from the typical cell-mediated to humoral immunity (Niciu et al., 2012). This shift can subsequently result in a weakened immune system, making the animal more susceptible to numerous pathogens (Calapre et al., 2013a). Heat stress has been shown to activate heat shock proteins (HSPs), which can promote cell proliferation and survival (Srikanth et al., 2017). Research found that HSPs are overexpressed in various cancers and have been implicated in carcinogenesis (Ciocca and Calderwood, 2005). Heat stress has also been documented to cause a change in animals’ adaptive immune function, transitioning from the typical cell-mediated to humoral immunity (Sophia et al., 2016). This shift can subsequently result in a weakened immune system, making the animal more susceptible to numerous pathogens (Vandana et al., 2019). Additionally, HS can lead to oxidative stress in various livestock, such as dairy cattle (Bernabucci et al., 2002), pigs (Liu et al., 2016), sheep (Chauhan et al., 2016), and poultry (Shakeri et al., 2019). This heightens their vulnerability to numerous pathogens and production-related illnesses.
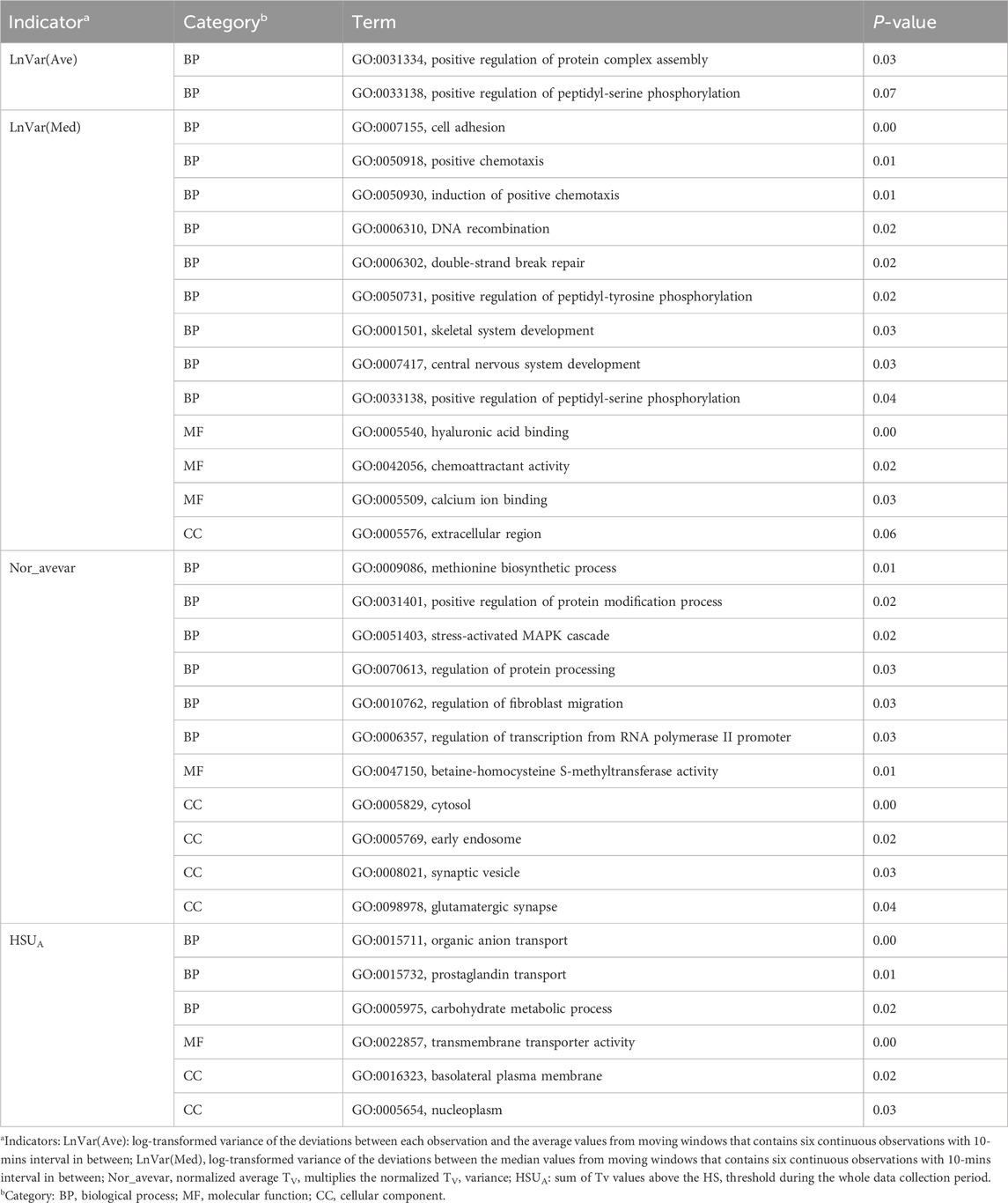
Table 2. Significantly enriched (P < 0.05) Gene Ontology (GO) terms identified for CR indicators.
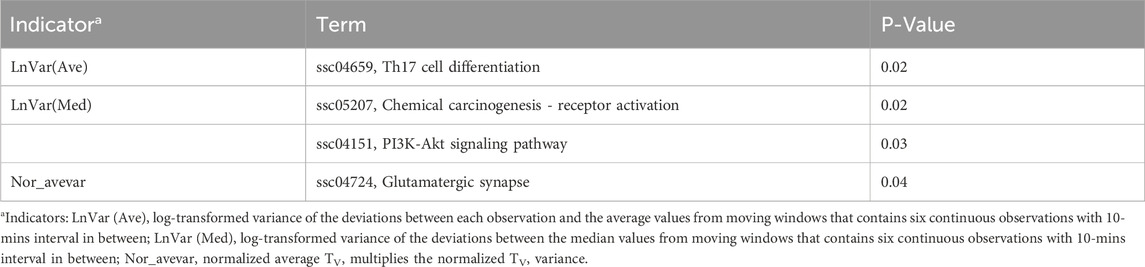
Table 3. Significantly enriched (P < 0.05) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways identified for CR indicators.
In addition, studies have shown that the immune responses in organisms are extremely sensitive to DNA damage that is caused by stressors (Nakad and Schumacher, 2016). The PI3K/AKT signaling pathway is involved in intracellular responses by reactive oxygen species (ROS) and inflammation caused by DNA fragmentation (Datta et al., 2023). Heat stress-induced testicular damage could be alleviated with melatonin, a potent antioxidant, in dairy goats by inhibiting the PI3K/AKT signaling pathway (Liu et al., 2022). Previous research has indicated that thermal stress leads to a reduction in glutamatergic synapse transmission (Popoli et al., 2012). Besides, glutamatergic synapses have been demonstrated to play roles in HSPs synthesis. This synthesis aids in repairing stress-induced synaptic protein damage and bolsters neuroprotective mechanisms (Kiang and Tsokos, 1998). Heat tolerance, defined as the rate of decline in milk production (slope traits) in response to a rising temperature–humidity index, is significantly associated with the enrichment of the glutamatergic synapse pathway in Holstein cows (Cheruiyot et al., 2021).
A total of 13, two, 17, eight, and one significant GO terms were enriched for Lnvar(Med), Lnvar(Ave), Nor_avevar, HSUA, and HSUB, respectively. The functions of enriched GO terms are similar to those of KEGG pathways. These GO terms are related to DNA damage and repair (e.g., DNA recombination, double-strand break repair), stress responses (e.g., stress-activated MAPK cascade), protein modifications (e.g., positive regulation of peptidyl-serine phosphorylation, positive regulation of peptidyl-tyrosine phosphorylation, and positive regulation of protein complex assembly), nervous system (e.g., central nervous system development, glutamatergic synapse, and synaptic vesicle), and cell structure and mechanics. Various types of DNA damage, including the induction of double-strand breaks in DNA (Ning et al., 2021; Habibi et al., 2022), are directly affected by HS. DNA recombination is one mechanism cells use to repair certain types of DNA damage. Previous research showed that the upregulated genes are mainly involved in DNA or protein damage/recombination, cell cycle processes, biogenesis, and stress and immune responses using transcriptome analysis in heat-stressed finishing pigs (Ma et al., 2019).
A substantial number of phosphorylation changes are induced by severe heat stress and occur with kinetics similar to the inhibition of protein synthesis (Duncan and Hershey, 1989). This has been evidenced by the detection of phosphorylation-related GO terms and functions in various species under HS conditions, including buffalos (Muthukumar et al., 2014), broilers (Kim et al., 2022), and swine (Yu et al., 2010; Cross et al., 2018). Notably, phosphorylation is crucial for the transcriptional activity of the heat shock transcription factor 1 and for triggering the heat shock response (Holmberg et al., 2001). Moreover, HS has been shown to activate MAPK phosphorylation in different cell types, such as intestinal cells, lung fibroblasts, and chondrocytes (Liu et al., 2019), and it has also been associated with cell and tissue injury (Banerjee Mustafi et al., 2009).
The genomic regions around candidate SNPs are shown to be linked with QTL regions associated with different traits. The major fraction of QTL annotated in this study belonged to the “Meat and Carcass” type, which accounts for 43.75% of the total QTL, and average daily gain and bone weight were the most traits that we enriched for Meat and Carcass QTL. Meanwhile, the genomic regions identified overlapped with several QTLs previously related to production, reproduction, health, and exterior traits (conformation and appearance), as shown in Table 4.
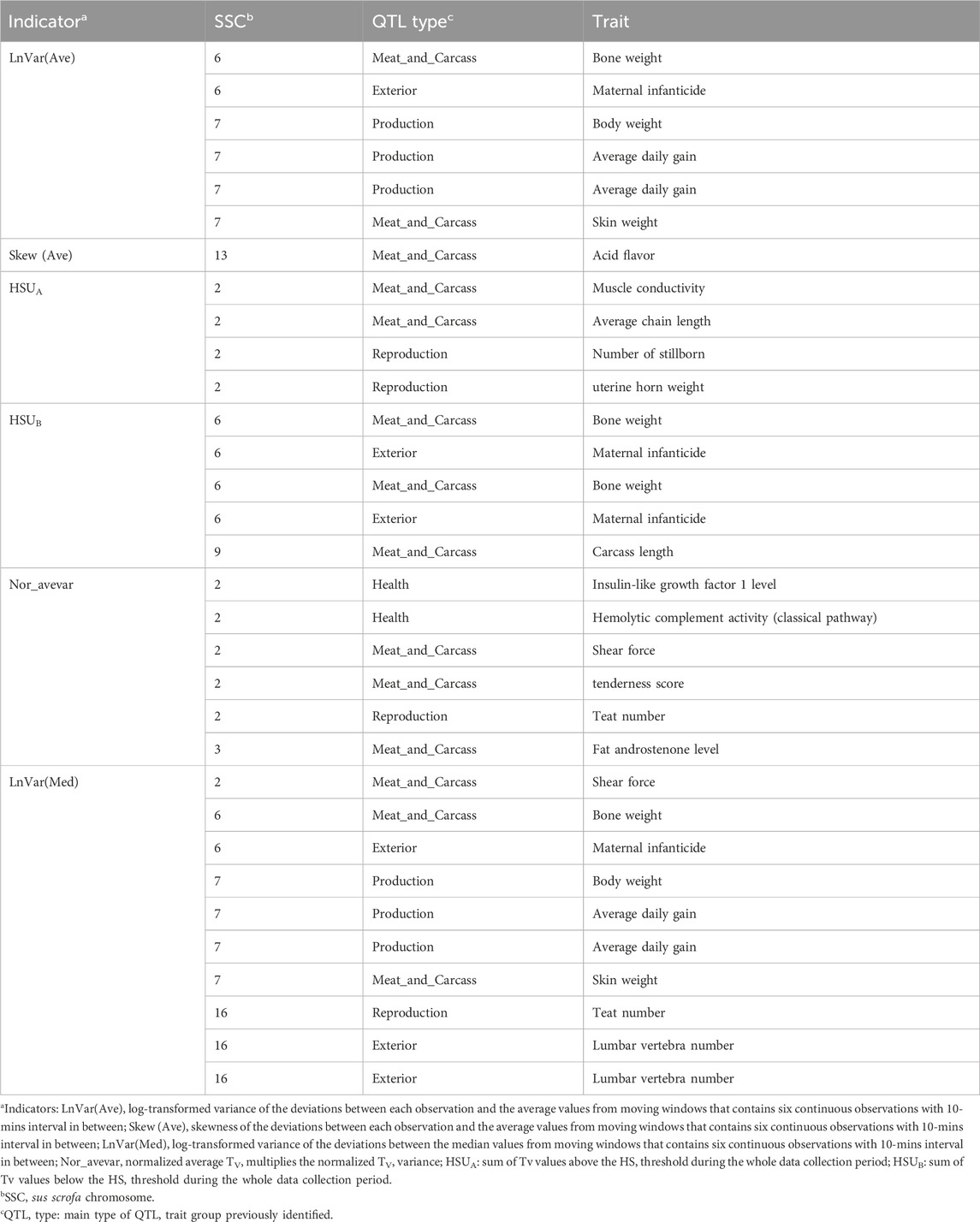
Table 4. Significantly enriched (P < 0.05) QTL identified for CR indicators.
This is the first GWAS for CR indicators derived from automatically measured TV. These significant SNPs hold great potential for enhancing genomic predictions for CR in pigs, by incorporating more SNPs located in the regions of these significant SNPs into existing commercial SNP panels to improve the prediction accuracy. Small sample size may limited the power of analysis, this analysis should be conducted in a larger population. Besides, different weights for these important SNPs or genes could be given through biology-driven genomic prediction methods [e.g., different subsets of SNPs were used for genomic predictions (Li et al., 2018)]. However, even though using different significance thresholds for GWAS, identifying the causal mutations for these CR indicators remains challenging due to the linkage disequilibrium. It would be better to use whole genome sequencing data to fully capture LD patterns, thereby achieving higher GWAS power compared to array-based GWAS (Pengelly et al., 2015). Thus, future research efforts should prioritize additional biological validations. In our study, we used a crossbred population. Similar analyses should be conducted in other populations with different genetic backgrounds to determine if the CR indicators are universally applicable.
4 ConclusionThe study focuses on sows and explores various CR indicators to better understand the genetic factors and biological mechanisms behind climatic resilience. We identified multiple genetic regions associated with different CR indicators, revealing that CR is a highly polygenic trait with small effect sizes distributed across the genome. Furthermore, many heat tolerance or HS related genes in our study, such as HSP90AB1, DMGDH, and HOMER1, have been identified. Additionally, the functional analyses showed the complexity of CR, involving various adaptive responses, from behavioral to cellular. These findings highlight the possibility of selecting more heat-tolerant individuals based on the identified SNP for CR indicators.
Data availability statementThe data analyzed in this study is subject to the following licenses/restrictions: The phenotypic and genomic data used in this study are a property of the industry partner that contributed to the study and therefore are not readily available due to its commercially sensitivity. Requests to access these datasets should be directed to the corresponding author (LB, britol@purdue.edu).
Ethics statementThe animal study was approved by Purdue University Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributionsHW: Conceptualization, Formal Analysis, Writing–original draft, Writing–review and editing. JJ: Writing–review and editing, Data curation. HM: Supervision, Writing–review and editing. AA: Supervision, Writing–review and editing. FD: Writing–review and editing. AR: Writing–review and editing. YH: Data curation, Resources, Writing–review and editing. FT: Writing–review and editing. CM: Writing–review and editing. AS: Writing–review and editing. LB: Conceptualization, Project administration, Supervision, Writing–review and editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Agriculture and Food Research Initiative Competitive Grant number 2022-67021-37022 from the USDA National Institute of Food and Agriculture.
AcknowledgmentsThe authors acknowledge Caitlin Wager, Sharlene O. Hartman, MaryKate Byrd, Jason R. Graham, Guadalupe Ceja, and Alexis Smith (Purdue University, West Lafayette, IN, United States), Nihya Alston and Dana Cinao (North Carolina State University, Raleigh, NC, United States), John Tyer, Jeremy Howard, Ashley E. DeDecker, Youping Gu, and Laurie Weston (Smithfield Foods, Warsaw, NC, United States) for their contributions to the study planning and data collection. We are also grateful to all the help provided by the Maple Hill farm employees during the data collection period and for the genomic datasets provided by Smithfield Premium Genetics (Raleigh, NC, United States).
Conflict of interestYH was employed by Smithfield Foods. The remaining authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare that they have no competing interests.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1498380/full#supplementary-material
ReferencesAhbara, A. M., Khbou, M. K., Rhomdhane, R., Sassi, L., Gharbi, M., Haile, A., et al. (2022). Genome variation in tick infestation and cryptic divergence in Tunisian indigenous sheep. BMC Genomics 23, 167. doi:10.1186/s12864-022-08321-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Alonso-Hearn, M., Badia-Bringué, G., and Canive, M. (2022). Genome-wide association studies for the identification of cattle susceptible and resilient to paratuberculosis. Front. Vet. Sci. 9, 935133. doi:10.3389/fvets.2022.935133
PubMed Abstract | CrossRef Full Text | Google Scholar
Álvarez, I., Fernández, I., Traoré, A., Pérez-Pardal, L., Menéndez, N. A., and Goyache, F. (2020). Genomic scan of selective sweeps in Djallonké (West African Dwarf) sheep shed light on adaptation to harsh environments. Sci. Rep. 10, 2824. doi:10.1038/s41598-020-59839-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi:10.1038/75556
PubMed Abstract | CrossRef Full Text | Google Scholar
Banerjee Mustafi, S., Chakraborty, P. K., Dey, R. S., and Raha, S. (2009). Heat stress upregulates chaperone heat shock protein 70 and antioxidant manganese superoxide dismutase through reactive oxygen species (ROS), p38MAPK, and Akt. Cell. Stress Chaperones 14, 579–589. doi:10.1007/s12192-009-0109-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Beemelmanns, A., Zanuzzo, F. S., Xue, X., Sandrelli, R. M., Rise, M. L., and Gamperl, A. K. (2021). The transcriptomic responses of Atlantic salmon (Salmo salar) to high temperature stress alone, and in combination with moderate hypoxia. BMC Genomics 22, 261. doi:10.1186/s12864-021-07464-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Bernabucci, U., Ronchi, B., Lacetera, N., and Nardone, A. (2002). Markers of oxidative status in plasma and erythrocytes of transition dairy cows during hot season. J. Dairy Sci. 85, 2173–2179. doi:10.3168/jds.S0022-0302(02)74296-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Braz, C. U., Rowan, T. N., Schnabel, R. D., and Decker, J. E. (2021). Genome-wide association analyses identify genotype-by-environment interactions of growth traits in Simmental cattle. Sci. Rep. 11, 13335. doi:10.1038/s41598-021-92455-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Cabezón, F. A., Schinckel, A. P., Richert, B. T., Peralta, W. A., and Gandarillas, M. (2017). Technical Note: application of models to estimate daily heat production of lactating sows. Prof. Animal Sci. 33, 357–362. doi:10.15232/pas.2016-01583
CrossRef Full Text | Google Scholar
Charoensook, R., Gatphayak, K., Sharifi, A. R., Chaisongkram, C., Brenig, B., and Knorr, C. (2012). Polymorphisms in the bovine HSP90AB1 gene are associated with heat tolerance in Thai indigenous cattle. Trop. Anim. Health Prod. 44, 921–928. doi:10.1007/s11250-011-9989-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Chauhan, S. S., Ponnampalam, E. N., Celi, P., Hopkins, D. L., Leury, B. J., and Dunshea, F. R. (2016). High dietary vitamin E and selenium improves feed intake and weight gain of finisher lambs and maintains redox homeostasis under hot conditions. Small Ruminant Res. 137, 17–23. doi:10.1016/j.smallrumres.2016.02.011
CrossRef Full Text | Google Scholar
Chen, S.-Y., Boerman, J. P., Gloria, L. S., Pedrosa, V. B., Doucette, J., and Brito, L. F. (2023). Genomic-based genetic parameters for resilience across lactations in North American Holstein cattle based on variability in daily milk yield records. J. Dairy Sci. 106, 4133–4146. doi:10.3168/jds.2022-22754
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, S.-Y., Gloria, L. S., Pedrosa, V. B., Doucette, J., Boerman, J. P., and Brito, L. F. (2024). Unraveling the genomic background of resilience based on variability in milk yield and milk production levels in North American Holstein cattle through genome-wide association study and Mendelian randomization analyses. J. Dairy Sci. 107, 1035–1053. doi:10.3168/jds.2023-23650
PubMed Abstract | CrossRef Full Text | Google Scholar
Cheruiyot, E. K., Haile-Mariam, M., Cocks, B. G., MacLeod, I. M., Xiang, R., and Pryce, J. E. (2021). New loci and neuronal pathways for resilience to heat stress in cattle. Sci. Rep. 11, 16619. doi:10.1038/s41598-021-95816-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Ciocca, D. R., and Calderwood, S. K. (2005). Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell. Stress Chaper 10, 86–103. doi:10.1379/CSC-99r.1
PubMed Abstract | CrossRef Full Text | Google Scholar
Colditz, I. G., and Hine, B. C. (2016). Resilience in farm animals: biology, management, breeding and implications for animal welfare. Anim. Prod. Sci. 56, 1961. doi:10.1071/AN15297
CrossRef Full Text | Google Scholar
Cottrell, J. J., Liu, F., Hung, A. T., DiGiacomo, K., Chauhan, S. S., Leury, B. J., et al. (2015). Nutritional strategi
留言 (0)