
記住我
Orofacial clefts (OFC), including cleft lip and cleft palate, are one of the most common birth defects with an approximate incidence of 1.7 per 1,000 live births, vary with ethics, territories, prenatal exposure as well as socioeconomic status (SES) (Clark et al., 2003; Mossey et al., 2009; Mossey and Modell, 2012). Clefts can be presented as isolated malformations or as part of the overlapping manifestations of syndromes with distinct entities. Understanding of lip and palate development is essential for the mechanogenesis, prevention, treatment, and prognosis of OFC. Despite the discrepancies in facial anatomies and tissue origins, vertebrates are highly conserved in the early craniofacial development, which relies on the precisely regulated outgrowth of five facial prominences–the frontonasal prominence (FnP) that develop into the medial nasal (MnP) and lateral nasal prominences (LnP), the bilaterally paired maxillary (MxP) and mandibular prominences (MdP), and their subsequent fusion (Jiang et al., 2006). These facial processes, including the palatal shelves, are covered by epithelial cells of ectodermal origin, with the underneath mesenchyme derived primarily from the neural crest (Jiang et al., 2006; Li et al., 2017). The cranial neural crest cells, a stem cell population that emigrated from the borders between non-neural ectoderm and the closing neural tube, migrate ventrally via well-defined routes and populate the mesenchyme of the facial prominences, where they eventually contribute to the formation of craniofacial bone and cartilage, connective tissues and peripheral neurons and glia (Roth et al., 2021). Despite the high versatility in generating a vast range of cell types, the neural crest cells proliferate, migrate and acquest cell fate according to intrinsic programming as well as to constantly changed extrinsic instructions, including reciprocal interactions between NCC and the adjacent cells as well as chemotactic cues in their microenvironment (Erickson et al., 2023). The surface ectoderm outlining embryonic facial prominences acts as the epithelial signaling center of morphogenic instructions, which are bidirectional processes as the expression of agonists/antagonists by neural crest cells are necessary for the maintenance of pattern of expression of morphogenic signaling molecules in the epithelium (Marchini et al., 2021). Proliferation and directed expansion of mesenchymal cells depend on signals from the epithelium, and signals from the mesenchyme influence epithelial cell development (Richman and Tickle, 1989; Francis-West et al., 1998; McGonnell et al., 1998). These interactions involve many intercellular signaling pathways and transcription factors, including sonic hedgehog (Shh), fibroblast growth factor (Fgf), bone morphogenetic protein (Bmp)/transforming growth factor-β (Tgf-β), wingless-type MMTV integration site (Wnt), retinoic acid (RA), and others (Jiang et al., 2006; Li et al., 2017; Atukorala and Ratnayake, 2021; Won et al., 2023).
There are numerous reviews on the craniofacial development and the pathogenesis of OFC from genetic and cellular perspectives (Reynolds et al., 2019; Reynolds et al., 2020; Hammond and Dixon, 2022). However, although EMI are essential processes underlying embryonic morphogenesis such as limb development, odontogenesis and organ formation, an updated comprehensive summary of the epithelial-mesenchymal crosstalk in regulating cranial neural crest cell differentiation and craniofacial development is not available. In this narrative review, after a brief overview of the early craniofacial development, we focused on the mutual communications between the epithelium and mesenchyme in respect to the fundamental cellular events, including NCC proliferation and migration, NCC differentiation, fusion, and epithelial-mesenchymal transition (EMT) during craniofacial formation as well as how disruption in these processes led to malformations.
2 Early craniofacial development and morphogenesisMammalian heads are distinctive covariation structures that are determined by integrated variations in fundamental developmental processes including neural crest migration/proliferation/patterning, facial process fusion, mesenchyme condensation/differentiation, cartilage/bone formation, brain growth and muscle-bone interaction, as well as the entire somatic development (Carlson and Byron, 2008). These processes arise in a developmental time-order from early after conception to adolescence. Despite the vast interspecies phenotypic differences, the early craniofacial development is highly conserved and for this reason, most of our knowledge about the molecular mechanisms have obtained from using mouse as one of the model organisms (Tamarin and Boyde, 1977; Gritli-Linde, 2008; 2012). Craniofacial morphogenesis starts approximately from the fourth week of human embryonic development (correspondent to E8.5 in murine embryogenesis) with the cranial neural crest cells (CNCC) delaminated from the edge of dorsal neural tube through the process of EMT (O'Rahilly and Müller, 2007; Minoux and Rijli, 2010; Simões-Costa and Bronner, 2015; Soldatov et al., 2019). These CNCC then migrate ventrally to populate the mesenchyme underneath the surface ectoderm of the frontonasal process and the 1st branchial arch, which develop into the maxillary and mandibular processes at fifth week of human and at E9.5 of mouse gestation, respectively (Yoshida et al., 2008; Simões-Costa and Bronner, 2015; Soldatov et al., 2019). By this stage, the facial primordium consisting of five separate epithelium-covered prominences surrounding the invaginated stomodeum can be easily observed, with the FnP at ventromedial, a pair of MxPs and a pair of MdPs at the lateral sides. At stage 14 (33 days after fertilization) of human embryogenesis, equivalent to E10.5 in mouse embryo, the FnP divides into paired MnPs and LnPs by invaginating nasal placodes as well as the proliferating primordial mesenchymal cells (Jiang et al., 2006). The close contact between approaching MnPs, LnPs and MxPs leads to their fusion at the lambdoidal junction at the nasolacrimal groove by stage 16 (E11.0 in mouse embryogenesis) (Jiang et al., 2006).The epithelial seam of opposing prominences eventually breakdown to ensure a confluent mesenchyme covered by a continuous sheath of epithelium by stage 19 (mouse E12.5) (Jiang et al., 2006; Gritli-Linde, 2008; Minoux and Rijli, 2010). The convergence of MnPs in the midline and their inward growth into oral cavity generate the primary palate, which fuses with the secondary palate developed from MxPs starting from approximately E11.5 in mouse (Yu et al., 2017). The palatal shelf grows bilaterally from the inner side of the MxP and extends antero-posteriorly along the lateral walls of the oropharynx. In mice, starting at E11.5, the palatal shelves flanking the tongue grow vertically in the oral-nasal cavity, followed by elevation to a horizontal position above the tongue within a narrow time frame between E14.0-E14.5 (Yu et al., 2017). Further polarized growth results in the approach of the contralateral palatal shelf, which then adheres along its medial edge epithelia (MEE) to form a medial epithelial seam (MES). The gradual disappearance of the MES allows the palatal shelf to fuse along the midline (Gritli-Linde, 2007; 2008). The synchronized growth and fusion of facial primordia are precisely controlled by both genetic and environmental regulations, and perturbations in which may result in abnormalities including orofacial clefts.
3 Epithelial-mesenchymal interaction in the specification, delamination and migration of CNCCsThe vertebrate unique neural crest cells (NCCs) arise along the entire length of neural tube at the border between neural and non-neural ectoderm, where they subjected to integrated signals from neural, non-neural ectoderm, and the mesoderm underneath (Dupin and Sommer, 2012). Induction of NCCs involves a complex gene regulatory network that works in a feed-forward manner. Signals including BMPs, WNTs, FGFs, and Notch drive the expression of the neural plate border specific transcription factors, which superimposed on the signaling ligands to induce subsequent expression of another set of neural crest specific transcription factors (Knecht and Bronner-Fraser, 2002). In chicken embryos, the expression of Bmp4 and Bmp7 in the non-neural ectoderm was activated and maintained by epidermal Notch (Endo et al., 2002). Addition of Bmp4 and Bmp7 induced neural crest cells in the explant culture of chicken intermediate neural plate in the absence of non-neural ectoderm in serum-containing nutrient rich media, but not in serum-free media (Liem et al., 1995; García-Castro et al., 2002). Therefore, diffusive Bmp gradient in the ectoderm and the expression of Bmp antagonists (Noggin, Cerberus, and Chordin, etc.) in the neural plate are precisely controlled to ensure proper levels of Bmps for the induction of the neural crest (Wilson et al., 1997; Marchant et al., 1998; Tribulo et al., 2003; Sauka-Spengler and Bronner-Fraser, 2008). Elevated Bmp signaling during NCC delamination and migration has also been demonstrated in chick embryos in recent research (Piacentino et al., 2021; Rekler and Kalcheim, 2022). Likewise, canonical Wnt signaling has been well-characterized in neural crest induction, migration, and EMT in multiple vertebrates, and has been thoroughly addressed in the review by Ji et al. (2019). Wnt signaling was shown to be necessary and sufficient to induce neural crest cells in multiple organisms both in vivo and in vitro, including in Xenopus embryo, in chick neuroepithelium explants, as well as in human embryonic cell culture (Jones and Trainor, 2004; Devotta et al., 2018; Gomez et al., 2019). In Xenopus embryos, Fgfs were strongly expressed in the paraxial mesoderm and was sufficient to induce the expression of a panel of neural crest markers in the ectoderm independent of Wnt, as demonstrated by injection of Fgf expression vector in vivo as well as in explants (Monsoro-Burq et al., 2003). In addition, Fgf signaling from the paraxial mesoderm was also shown in chick embryo to be necessary to prevent premature specification of the caudal NCC, and thereby, prevent subsequent EMT and migration (Martínez-Morales et al., 2011; Rekler and Kalcheim, 2022).
4 Epithelial-mesenchymal interaction in regulating facial primordial growthAccumulating evidence from experimental data using various animal models suggested an instructive role of epithelial signals in regulating the mesenchymal proliferation during facial primordial outgrowth. A region called frontonasal ectodermal zone (FEZ) in the frontonasal epithelium, specified by the dorsal-ventral molecular boundary of the expression of Fgf8/Shh, was first defined in stage 20 avian embryo and served as the signaling center to regulate the growth and patterning of facial primordia (Hu et al., 2003). A conserved molecular pattern of dorsal-ventral Fgf8/Shh expression was also found in mouse FEZ, which was presented as two separate signaling domains with discontinuous Shh expression on the left and right ventral side of the mouse medial nasal processes (Hu and Marcucio, 2009b; a). The bilateral pattern of Shh expression is also presented in human embryos in this region, indicating the potential fundamental differences in patterning information that distinguish mammalian faces (Odent et al., 1999). Hedgehog signaling is essential for craniofacial development, and among the three hedgehog enantiomers possessed by vertebrates, Shh has the broadest function in facial development (Abramyan, 2019). Shh is expressed in the facial primitive epithelium at approximately E11 in mice, and prior to its expression, facial mesenchymal cells have received Shh ligands, which possibly from the ventral neural tube epithelium, to activate the Shh signaling pathway (Kurosaka, 2015). In mouse embryos at stages between E9.5-E11.5, Fgf family members are also expressed in the facial primordium, where the ligands Fgf8, Fgf9, and Fgf10 are expressed in the facial ectoderm while Fgfr1 and Fgfr2 receptors are expressed in both facial ectoderm and mesenchyme (Bachler and Neubüser, 2001).
Using chick models, studies of ectopic transplantation of the FEZ showed the ectopic expression of the downstream genes in the facial mesenchyme and induced the subsequent alterations in patterning of morphogenesis (Hu et al., 2003). Shh induced expression of targets downstream of the Shh pathway (Ptc1 and Gli1) in the neighboring mesenchyme, while the expression of downstream targets of Fgf, Msx1 and AP2, was upregulated in the mesenchyme underneath the Fgf8-positive dorsal ectoderm even when Fgf signaling started fading at stage 25 (Hu and Helms, 1999; Hu et al., 2003). Shh can also regulate mesenchymal proliferation by modulating the forkhead box (Fox) genes (Foxf1/2), and both excessive and inadequate Shh signaling can lead to abnormal facial development (Hu and Helms, 1999; Jeong et al., 2004; Everson et al., 2017).
It is worth noting that the establishment of the molecular pattern of Fgf8/Shh in FEZ also relies on the regulation of other signaling pathways. For instance, eliminating canonical Wnt/β-catenin signaling in mouse facial ectoderm dramatically abolished Fgf8 expression while induced a ventrally expanded continuous medio-lateral expression zone of Shh and resulted in a deformity characterized by a narrow and pointed face, resembling that was observed in avian (Reid et al., 2011). On the contrary, sustained Wnt/β-catenin signaling led to upregulated and expanded Fgf8 expression in the entire facial ectoderm with restricted Shh expression in the most lateral region of FEZ, with no distinguishable craniofacial structures developed by E12.5 (Reid et al., 2011; Wang et al., 2011b). On the other hand, mesenchymal signaling also affect the molecular pattern in FEZ (Foppiano et al., 2007; Reid et al., 2011; Hu et al., 2015). Ectopic expression of Shh in the forebrain mesenchyme in chick embryos before facial development led to a murine-like Shh expression pattern in FEZ and a broader and shorter upper jaw resembling mouse embryo at later developmental stages (Hu and Marcucio, 2009a). In Wnt1Cre; Ptch1c/c mutant mouse embryos, downregulation of the expression of Ptch1, the major negative regulator/receptor of Shh, in the NCC-derived mesenchyme, resulted in aberrant upregulation of Shh signaling in the facial mesenchyme and loss-of-expression of Fgf8 in the frontonasal epithelium (Metzis et al., 2013).
The Bmp signaling pathway regulates a series of cell proliferation, apoptosis, and differentiation processes (Wan and Cao, 2005). Upon ligand binding, Bmps activate type I and type II receptor heterodimers and in turn activate transcriptional coactivators Smads, which lead to downstream target gene expression (Wang et al., 2014). Both Bmps and Bmp antagonists have been found expressed in the epithelium as well as in the mesenchyme of facial prominences (Trousse et al., 2001). Bmp2 and Bmp4 are expressed in a dynamic pattern in the chick embryo facial blastoderm. Bmp4 first expressed at HH16-17 in the facial ectoderm, and then sequentially in the mesenchyme of LnP, MnP, MxP and MdP while Bmp2 expressed in both epithelium and mesenchyme of the facial primordium (Francis-West et al., 1994). Implantation of Noggin-soaked beads in the FnP mesenchyme at HH15/16 or infection of neural crest cells at HH10 with a replication-competent retrovirus encoding the Bmp antagonist Noggin (RCAS-Noggin) abolished Shh expression in the ventral domains of FEZ (Foppiano et al., 2007; Hu et al., 2015). Ectodermal Shh also affects the expression of Bmps in the mesenchyme, and activation of the Shh signaling pathway in FnP increases the expression of Bmp2, Bmp4, and Bmp7 in its mesenchyme (Hu et al., 2015). Mutual antagonistical regulation was also observed between Bmp and Fgf8 signaling (Shigetani et al., 2000). Tp63 encodes transcription factors that belong to p53 family and regulate the transcription of members of multiple signaling pathways, including Notch, Wnt, Tgf-β and Bmps (Yang et al., 2006). Tp63 is widely expressed in the developing facial epithelium and loss-of-function in Tp63 has been found to cause developmental malformations including OFCs in both human and mice (Thomason et al., 2008). Elevated Bmp4 expression was shown in the epithelium of caudal LnP and MxP in Tp63 null mice from E10.5 to E11.5, whereas the expression of Fgf8 was absent in the overlapping region (Thomason et al., 2008). Tp63−/− mice displayed fully penetrating bilateral cleft lips due to the aberrant signaling of Bmp4 and Fgf8, which was the major positive regulator of the mesenchymal cell proliferation (Thomason et al., 2008). Correlations between the expression of the homeobox genes, Msx1 and Msx2, as well as the expression of the T-box transcription factor, Tbx22, in the developing facial primordia and Bmp signaling have been demonstrated in both chick and mouse embryos (Barlow and Francis-West, 1997; Jiang et al., 2006; Higashihori et al., 2010). Implantation of the antagonistic Noggin beads in FnP mesenchyme before lip fusion (HH22) downregulated Msx1/Msx2 expression in the epithelium and mesenchyme while strongly induced the mesenchymal expression of Tbx22, resulted in significantly inhibited mesenchymal proliferation (Ashique et al., 2002; Higashihori et al., 2010). Tbx22 expression in facial mesenchyme was also induced by ectodermal Fgf8 and was likely to act upstream of Msx, given that gain-of-function of Tbx22 reduced the expression of Msx2 and Dlx5 and resulted in craniofacial malformations including OFC (Fuchs et al., 2010; Higashihori et al., 2010). Enhanced Bmp signaling in the mesenchyme induced apoptosis of epithelial and mesenchymal cells and inhibited the expression of Fgf8 and Shh in the epithelium (Ashique et al., 2002). Collectively, a role for Bmp signaling in promoting mesenchymal proliferation was supported by most of the experimental data while its function in facial ectoderm was still unclear.
Both expression analysis and association studies indicate that Wnt signaling molecules including ligands, receptors/co-receptors and downstream effectors are involved in crosstalk with other morphogenic pathways and play crucial roles in facial development (Chiquet et al., 2008; Mostowska et al., 2012; Fontoura et al., 2015; Lu et al., 2015). Mutations in genes encoding a wide range of Wnt signaling components, including both β-catenin-dependent canonical and β-catenin-independent non-canonical pathways, have been revealed in association with OFC in humans and animal models (Reynolds et al., 2019). Precisely regulated Wnt signaling is essential for the maintenance of the proper gene expression pattern in facial ectoderm as well as the mesenchymal proliferation, demonstrated by the resultant craniofacial defects of both ectodermal conditional loss-of-function (LOF) and gain-of-function (GOF) mutations of β-catenin in mice (Reid et al., 2011; Wang et al., 2011b). The expression of multiple Fgf ligands (Fgf8, Fgf17, Fgf3) was downregulated in the β-catenin-LOF mice while the expression of Fgf8 was expanded and upregulated in the facial ectoderm in β-catenin-GOF mice (Reid et al., 2011; Wang et al., 2011b). Altered expression pattern of Shh was also observed in these mice, indicating the ectodermal Wnt signaling was a player in establishing the molecular pattern in FEZ. In mouse embryos, the expression of Wnt3 and Wnt9b, both activate β-catenin mediated canonical pathway, was detected in the distal regions of the surface ectoderm of FnP and 1st branchial arch derived structures as early as E9.5 (Lan et al., 2006). However, unlike Wnt9b, high expression of which was detected in the pre-fusion ectoderm and persistent in the epithelial seam after fusion at E11.5, the expression of Wnt3a was absent from the epithelial contacts in the pre-fusion zone. Loss-of-function mutations in Wnt3 cause craniofacial defects in human and early embryonic death in mice (Liu et al., 1999). No coding mutations in Wnt9b has been reported in human while Wnt9b null mice develop incomplete penetrance of bilateral cleft lip (Jin et al., 2012). Wnt9b/β-catenin induces mesenchymal cell proliferation by regulating the expression of Fgf8, Fgf10 and Fgf17 in the ectoderm (Jin et al., 2012). Another study showed that Msx1/Msx2 were also downstream targets of the Wnt/β-catenin signaling pathway during lip formation and fusion and their expression were downregulated in the facial mesenchyme of Wnt9b −/− mice (Song et al., 2009; Jin et al., 2012). Interestingly, although the expression of Bmp4 remained unaffected in either Wnt9b −/− or β-catenin-LOF mouse embryos, tissue specific knockout of Gpr177 (also known as Wntless, responsible for Wnt sorting/secretion) in the facial ectoderm (Foxg1-Cre; Gpr177) from E9.5 reduced Bmp4 expression in the facial epithelium and mesenchyme, indicating a role for non-canonical Wnt signaling in regulating Bmp4 expression (Reid et al., 2011; Wang et al., 2011b; Jin et al., 2012; Zhu et al., 2016).
Retinoid acid, one of the metabolites of vitamin A processed by sequential enzymatic reactions involving retinol dehydrogenases and aldehyde dehydrogenases, is another signaling molecule produced in FnP ectoderm (Duester, 2000). Depends on the duration and time of commencement of induction, both excessive and deficient RA signaling lead to craniofacial defects (Emmanouil-Nikoloussi et al., 2000; Dupé and Pellerin, 2009). Aldh6 is expressed in a restricted region in ventral epithelium of presumable FnP in chick embryos as early as at HH10-HH12 (Schneider et al., 2001). Expression of retinoid receptors including RARα, RXRβ, and RXRγ can be detected in the adjacent mesenchymal neural crest cells in the facial primordium (Rowe et al., 1992; Rowe and Brickell, 1995; Hoover and Glover, 1998). Transiently inhibition of the mesenchymal retinoid receptors by implanting RAR/RXR antagonists-soaked beads in the rostral head at HH10 resulted in loss of the expression of Fgf8/Shh in FnP epithelium, which in turn, led to reduced mesenchymal proliferation and increased apoptosis in the FnP mesenchyme (Schneider et al., 2001). On the other hand, RA signaling was subjected to negative regulation by Wnt, shown by the expanded expression of Raldh3 in the pre-fusion epithelium of the upper lip/nasal primordia in the Lrp6−/− mice that displayed inactivated Wnt signaling and full penetrance of cleft lip/palate (Song et al., 2009) (Figure 1).
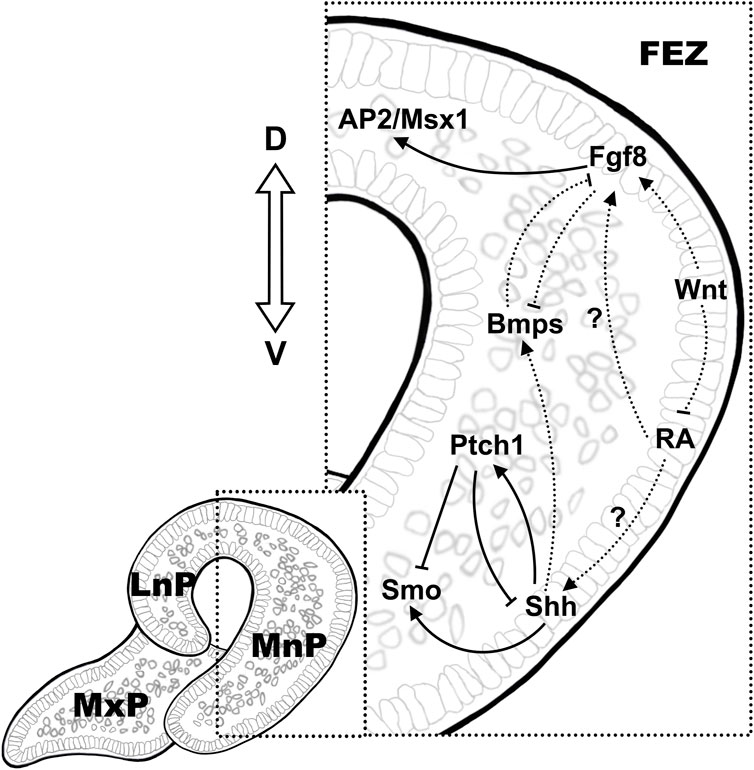
Figure 1. Signaling mechanisms in the establishing the molecular pattern in FEZ. The dorsal-ventral expression of Fgf8/Shh is conserved in the epithelium in FEZ, which is the signaling center in regulating mesenchymal proliferation during early facial development. Crosstalk between pathways is illustrated using solid lines and dashed lines, indicating direct and indirect regulations, respectively.
5 Epithelial-mesenchymal interaction in regulating palatal developmentThe Fgf-Shh signaling network involved in epithelial-mesenchymal interactions has also been shown to regulated palate development in mouse (Rice et al., 2004; Lan and Jiang, 2009). Embryonic palatogenesis displayed anteroposterior polarity with anterior bony hard palate and posterior muscular soft palate as well as oronasal polarity before fusion (Bush and Jiang, 2012). During palatal development (E12-14), the Fgf10 ligand is preferentially expressed in oral region of the anterior palatal mesenchyme adjacent to the epithelium while the receptor Fgfr2b is expressed in the palatal epithelium (Rice et al., 2004; Teshima et al., 2016). Both Fgfr2b−/− and Fgf10−/− mice have cleft palate and reduced proliferation of palatal shelf cells (Rice et al., 2004). The expression of Shh is restricted to the palatal epithelium on the oral side from E11.5 to E15.5, with the receptor Ptch1 and downstream transcription factor, Gli1, express in both palatal epithelium and mesenchyme (Ingham and McMahon, 2001; Lan and Jiang, 2009; Economou et al., 2012). Upon ligand binding, Ptch1 releases Smo from inhibition, which in turn activates GliFL and promotes target gene expression (Cohen et al., 2015). Epithelial Shh signal also regulated the palatal mesenchymal cell proliferation by modulating the expression of cycle D1/2 (Ccnd1/2), which was essential for palatal shelf growth (Lan and Jiang, 2009). Fgf10-Fgfr2b maintains Shh expression in palatal epithelium and vice versa, disrupted Shh signaling resulted in downregulation of Fgf10 in in palatal bone mesenchyme (Rice et al., 2004; Lan and Jiang, 2009). Odd-jump-related 2 (Osr2), homolog of Drosophila Odd-skipped, encoding a zinc finger protein of transcription factor that expressed in mouse embryos as early as E9.25 in the mesonephros, regulates pattern formation in a wide range of embryonic structures (Lan et al., 2001). Osr2 was highly expressed in the mesenchyme of palate shelves at E12.5 and the expression were decreased by E14.5 (Lan et al., 2001). Osr2−/− mouse embryos displayed cleft palate and reduced Fgf10 expression in the palatal mesenchyme (Zhou et al., 2013). In Osr2-Cre; Smoc/c mouse embryos in vivo as well as in mouse palate shelf cultures in vitro, dysregulation in Shh-Smo signaling also altered Bmp2/Bmp4 expression in the underlying mesenchyme in the anterior palate, upregulated Bmp2 expression induced by exogenous Shh while reduced Bmp2 expression and increased Bmp4 expression in Osr2-Cre; Smoc/c mouse palate shelves (Lan and Jiang, 2009).
Another member of the Fgf family, Fgf7, is nevertheless expressed in the nasal region of the palatal mesenchyme and is regulated by Dlx5, which displayed overlapping expression pattern (Han et al., 2009). The expression of Fgf7 in the oral region of palate mesenchyme was repressed by Shh. Loss-of-function of Dlx5 resulted in downregulation of Fgf7 and the expansion of Shh to the nasal side of palatal epithelium in Dlx−/− mice that displayed cleft palates with reduced proliferation of mesenchymal cells (Han et al., 2009). However, excessive Shh signaling in in palatal epithelium also caused cleft palate (Cobourne et al., 2009). Therefore, it was observed that the palatal fusion defects in palato-mesenchymal proliferation-deficient (Msx−/−) mice were rescued by the expanded Shh expression in Msx1−/−Dlx5−/− double knockout mice (Han et al., 2009). Inhibition of Shh expression by expression of Fgf18 and Fgf8 in palate mesenchyme has also been shown in mice. The expression of Shh in palate epithelium was significantly downregulated in conditional knockout mice of Foxf1/2, which displayed ectopic overexpression of Fgf18 in the oral side of palatal mesenchyme while addition of exogenous Fgf18 protein to cultured palatal explants suppressed Shh expression in the palatal epithelium (Xu et al., 2016). Activation of ectopic Fgf8 expression in the palatal mesenchyme in Osr2-CreKI; Rosa26R-Fgf8 mice resulted in dramatically increased proliferation of posterior palatal mesenchymal cells whereas decreased epithelial cell proliferation due to downregulated Shh expression (Wu et al., 2015). Further investigations suggested the inhibition of Shh expression in the palate epithelium by Fgf8 was secondary, likely to be the outcome of disrupted Shh-Fgf10 feedback loop (Wu et al., 2015). In addition, ectopic activation of Fgf8 also suppressed the expression of Dlx5 and Fgf7 (Wu et al., 2015). Therefore, the altered expression of Fgf8 in mouse embryonic palate may affect cell proliferation via a combined mechanisms involving multiple EMI pathways.
Bmp signaling is critical in the regulation of palatal growth and mesenchymal condensation/ossification (Parada and Chai, 2012). In mouse palatogenesis, the expression of Bmp4 was observed in the palatal epithelium and mesenchyme at E12.5 and was restricted to the anterior palatal mesenchyme at E13.5, while the expression of Bmp2 was observed in the epithelium and mesenchyme of the anterior region of the palatal shelf (Zhang et al., 2002). Inactivation of Shh signaling in the palatal mesenchyme by tissue specific knockout of Smo in the palatal mesenchyme (Osr2-IresCre; Smoc/c) led to overexpression of Bmp4 and Msx1 while led to downregulation of Bmp2 (Lan and Jiang, 2009). However, proper Bmp4 signaling is also essential for maintaining Shh expression in the medial edge epithelium, evidenced by the restored Shh and Bmp2 expression as well as the cell proliferation in Msx−/− mice by ectopic Bmp4 expression (Zhang et al., 2002). Given that Bmp4 was shown to act downstream of Msx1, the induced Msx1 expression in palatal explant cultures by implantation of Bmp4-soaked beads might be the consequence of the inhibited Shh expression due to the feedback regulation between Shh and Bmp4 (Chen et al., 1996; Zhang et al., 2002; Hilliard et al., 2005). The Bmp antagonist Noggin is also expressed in the palatal epithelium (He et al., 2010). Noggin−/− mouse embryos displayed defective proliferation and excessive apoptosis in the mesenchyme of palatal shelves. Reduced Smad1/5/8 phosphorylation in the anterior palatal mesenchyme and upregulation of Smad1/5/8 along the A-P axis in the posterior palatal mesenchyme have been demonstrated in these mouse embryos, whereas Bmp2 is reduced in the anterior palatal mesenchyme and ectopically expressed in the posterior palatal oral side of the epithelium (He et al., 2010). The altered expression of Bmp2 along the A-P axis in Noggin−/− mutants might be attributed in part to the changes in the levels of Smad1/5/8 phosphorylation in the palatal mesenchyme. Conditional knockout of the receptor Bmpr1a in the palatal mesenchyme and epithelium in mice (Nestin-Cre; Bmpr1af/-) results in cleft lip and cleft palate (Liu et al., 2005). Further knockout studies involving tissue specific inactivation of Bmpr1a in mouse palatal mesenchyme (Osr2-IresCre; Bmpr1af/f) showed that the expression of Shh and Msx1/Fgf10 was downregulated in the anterior palatal epithelium and mesenchyme, respectively, while the expression of Bmp4 and Bmp2 was increased in compensation in the palatal mesenchyme (Baek et al., 2011). These mice displayed cleft in anterior secondary palate due to reduced cell proliferation. However, deletion of Bmpr1a expression in the palatal epithelium only (K14-Cre; Bmpr1af/-) resulted in normal palatal development (Andl et al., 2004).
The expression of Gpr177 is found in both palatal epithelium and mesenchyme and promotes Wnt ligands secretion (Bänziger et al., 2006; Liu et al., 2015). Elimination of Wnt signaling by conditional knockout of Gpr177 in NCC-derived mesenchyme (Gpr177; Wnt1-Cre) resulted in abnormal cell proliferation and increased cell death in the palatal shelf, accompanied with upregulated expression of Msx1 in the anterior palatal mesenchyme and increased palatal epithelial Shh expression (Liu et al., 2015). Similar to its obligatory roles in establishing Fgf/Shh expression pattern in FEZ, activated Wnt signaling and high expression of β-catenin was also observed in the palatal rugae epithelium, which served as the Shh signaling center in palatal development (Lin et al., 2011). Conditional inactivation of β-catenin in the palatal epithelium at the beginning of palatal development at E12.5 in mouse embryos ablated Shh expression in the palatal rugae and consequently, disrupted the A-P expansion of the palatal rugae and palatine bone development (Lin et al., 2011). Wnt5a was expressed in gradience along the A-P axis in the mesenchyme during mouse palatogenesis (He et al., 2008). Downregulated Bmp4 expression in the mesenchyme and Shh in MEE of the anterior palate whereas ectopic Bmp4 expression in the mesenchyme and Shh expression in the nasal side epithelium of the posterior palate were observed in E13.5 Wnt5a−/− mouse embryos, which displayed complete cleft palate (He et al., 2008). The expression of Shh receptor, Ptch1, and the downstream target of Bmp4, Msx1, changed accordingly in the developing palate of Wnt5a−/− mouse embryos (He et al., 2008). These observations were in line with the epistatic effect of Wnt signaling in directing the establishment of the molecular pattern during upper lip formation. Null mutation in the Wnt5a receptor, Ror2, resulted in similar alterations in the gene expression in mouse embryonic palate and overlapping phenotype, indicating a non-canonical Wnt pathway in the regulation of palatogenesis.
Enzymes of cytochrome P450 subfamily 26 (CYP26) play dominant roles in RA degradation and display distinctive expression pattern in embryonic palate, with restricted expression of Cyp26a1 in the epithelium and Cyp26b1 in the mesenchyme (White et al., 1997; Okano et al., 2012). Cyp26b1−/− mouse embryos suffered from cleft palate, displaying downregulated expression of Fgf10 and Bmp2 in the palatal mesenchyme and downregulated Tbx1 in the palatal epithelium, respectively (Okano et al., 2012). Both the expression of Cyp26a1 and Cyp26b1 in the palatal shelf was reduced in ShhCreERT2/Shhf mutant mice, suggested an epistatic inhibitory effect of epithelial Shh signaling in regulating mesenchymal and epithelial RA signaling (El Shahawy et al., 2019). Antagonistic interaction was also found between RA signaling and Tgfβ pathway in regulating palatogenesis. Exogenous RA induced apoptosis in palatal mesenchymal cells and prevented palatal shelf fusion by inhibiting the Tgfβ-Smad pathway in the MES and palatal mesenchyme, and vice versa (Yu et al., 2005; Yu and Xing, 2006; Wang et al., 2011a; Liu et al., 2014).
In addition, these aforementioned signaling pathways are integrated to regulate the composition of the extracellular matrix, which served as the fundamental infrastructure for cell adhesion to guide the directional growth and elevation of embryonic palate (Wang et al., 2020). For instance, Collagens (Col), the major component of the extracellular matrix (ECM), are expressed extensively in the palatal mesenchyme before and after the elevation of the PS (Silver et al., 1981). The forkhead transcription factor, Foxf2, induced by ectodermal Shh and expressed in the cranial neural crest cells in MnP/MdP, activate canonical Tgfβ signaling in the palatal mesenchyme (Nik et al., 2016; Xu et al., 2016). The reduced expression of Tgf-β2 resulted in mesenchymal hypoplasia together with reduced production of Col I and several other extracellular proteins in Foxf2−/− mouse embryos that displayed cleft secondary palate (Nik et al., 2016). In contrast, the other two Tgfβ ligands, Tgfβ1 and Tgf-β3 are expressed in the palatal epithelium including MEE (Fitzpatrick et al., 1990). Tgf-β1 induced the proliferation and Col I and III synthesis in cultured palatal mesenchymal cells in vitro (Li et al., 2012). Downregulation of Tgf-β3 reduced Shh expression throughout E12.5-E15.5 in palate rugae and resulted in the failure of palate fusion without significant reduction in palatal growth (Sasaki et al., 2007).
Mutual regulatory mechanisms are also commonly recognized between the signaling pathways and the transcription factors. The basic helix-loop-helix (bHLH) transcription factors dHAND/HAND2 is expressed in cephalic neural crest cells as early as at E9.5 just after migration, as demonstrated using the dHAND-lacZ transgenic mice (Massari and Murre, 2000; Ruest et al., 2003; Barbosa et al., 2007). At later developmental stages, expression of Hand2 is found in the anterior palatal mesenchyme and MEE and the expression was ablated in Osr2-Cre; pMes-Nog mice that had attenuated Bmp signaling in the mesenchyme (Xiong et al., 2009). Inactivation of Hand2 in the palatal mesenchyme had no effect on palatal development while elimination of Hand2 in the palatal epithelium led to cleft palate resulted from premature death of periderm cells in the MEE and reduced mesenchymal proliferation via modulating Shh expression (Morikawa et al., 2007; Xiong et al., 2009). Early expression of the paired-box transcription factor, Pax9, in the palate region, became detectable at E12.5 in the mesenchyme in a posterior-anterior gradient pattern as well as in the posterior epithelium (Zhou et al., 2013). This was followed by a gradual decrease of expression in the mesenchyme, accompanied by intensely confined expression in the MEE during palate fusion (E14.5-E15.5) (Sasaki et al., 2007). The mutant mice exhibit cleft palate and die shortly after birth (Kist et al., 2007). The expression of Pax9 was regulated by epithelial Fgf signaling and exogenous Fgf8 induced Pax9 expression in the posterior palatal mesenchyme (Hilliard et al., 2005). The expression of Shh in the palatal rugae as well as the expression of Bmp4, Fgf10, Msx1 and Osr2 in the palatal mesenchyme were significantly downregulated in Pax9 null mice, suggesting Pax9 as a core player in converting the signaling cues to mediate the communication between epithelium and mesenchyme (Zhou et al., 2013) (Figure 2).
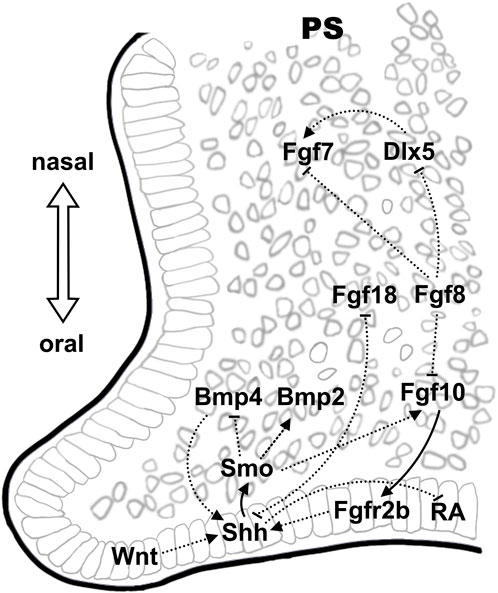
Figure 2. Epithelial-mesenchymal signaling interactions during palate development. The Shh-Fgf signaling network, which display oronasal polarity, is also found critical in palatogenesis. Wnt signaling in the epithelium on the oral side demonstrate an epistatic effect in regulating Shh expression. Crosstalk between pathways is illustrated using solid lines and dashed lines, indicating direct and indirect regulations, respectively.
6 Epithelial-mesenchymal interaction in the processes of lip and palate fusionThe growth and expansion of the facial prominences eventually lead to their fusion in the midline, which is a process involving epithelial seam formation followed by EMT and/or epithelial regression (Gaare and Langman, 1977; Sun et al., 2000). Fusion is a sequential and directional process that has species differences, with fusion initiated between MnP and LnP in posterior-anterior direction followed by the fusion between MnP and MxP in mice (Jiang et al., 2006). Wnt9b is expressed in the epithelial seam between the fused MnP and LnP (Lan et al., 2006). Meeting of MnP, LnP and MxP create a three-way seam called lambdoidal junction, where Wnt-p63-Irf6 are intensively expressed to maintain apoptosis in epithelial suture cells (Ferretti et al., 2011). The Wnt-p63-Irf6 regulatory module was disrupted in Ptch1wiggable mice embryos that was shown to have enhanced Shh signaling in the mesenchyme (Kurosaka et al., 2014). Further investigations showed that multiple canonical Wnt inhibitors’, such as Vax1, Sfrps and Frzb, were upregulated while the expression of the transcription factor of Irf6 promotor, Tfap2a, was downregulated in the mesenchyme of facial processes of Ptch1 wiggable mice (Kurosaka et al., 2014).
The periderm is a barrier that prevents any unwanted adhesion of the palatal shelf to the surrounding tissues and the failure of periosteum differentiation leads to Cleft Lip and Palate (CLP) (Hammond et al., 2019). Expression of Irf6 and its downstream target, the receptor-interacting protein kinase 4 (RIPK4), was observed throughout mouse palatogenesis in the palatal epithelium and was important for peridermal formation (De Groote et al., 2015; Kousa and Schutte, 2016). Mutations in these genes resulted in anomalies including cleft palate as well as abnormal adhesion of the palatal shelves to the mandible and/or tongue due to impaired peridermal differentiation and ectopic apical E-cadherin distribution, respectively (Ingraham et al., 2006; De Groote et al., 2015). Jag2, one of the surface ligands of the receptors of Notch family, mediates the process of differentiation in the oral periderm and the mutant mice displayed CLP with palate-tongue fusion (Shawber et al., 1996). In mice, high expression of Jag2 was observed in the tongue, mandibular and maxillary epithelia from E12.5 to E14.5, with its receptor, Notch1 expressed in overlapping regions with a time delay at E13.5 (Casey et al., 2006). Downregulated Jag2 expression in the oral epithelia was observed in Fgf10−/− mutant mice that displayed abnormal palate-tongue fusion and likewise, although Fgf10 expression was unaltered in the mesenchyme, reduced Fgfr2b expression in the oral epithelium was observed in Jag2 mutant mice, indicating that the Fgf signaling in the mesenchyme might regulate the epithelial differentiation by a feedback regulation of Jag2-Notch signaling (Alappat et al., 2005). The transcription factor Tbx1 that is expressed in epithelial cells of early facial processes has also been found expressed in palatal epithelium in mice from E12.5 to E15.5 (Funato et al., 2012). The results of expression analysis suggested altered gene expression of Fgf10, Bmp4 and Pax9, etc., in the palatal mesenchyme of Tbx1−/− mice that showed cleft palate with abnormal epithelial adhesions between the palatal shelf and the mandible (Goudy et al., 2010; Funato et al., 2012). These findings may partly explain the defective mesenchymal proliferation as well as impaired epithelium differentiation observed during palatogenesis in Tbx1-null mice (Funato et al., 2012; Zoupa et al., 2018).
The growth and elevation of palatal shelves lead to their fusion in the midline above the tongue. The epithelial cells in the medial edge epithelia are subjected to three different fates, programmed cell death, migration or EMT, fail in which resulted in submucous cleft (Jin and Ding, 2006). Wnt11 is strongly expressed in the MEE region and Fgfr1b is expressed in the palatal mesenchyme (Lee et al., 2008). The mutual inhibitory regulation between Wnt11 and Fgfr1b was shown to be critical for the apoptosis during fusion in cultured palatal shelves in vitro (Lee et al., 2008). Initial palatal shelf contact is mediated by chondroitin sulfate proteoglycans expressed on filamentous pseudopods that formed on the apical surface of periderm cells, followed by migration of periderm cells out of the MES (Taya et al., 1999; Martínez-Alvarez et al., 2000; Cuervo and Covarrubias, 2004; Vaziri Sani et al., 2010; Richardson et al., 2017). Tgf-β3 is expressed in MEE and knockdown of Tgf-β3 or epithelial-specific deletion of Tgfbr1 or Tgfbr2 results in impaired MEE degeneration and palatal fusion (Proetzel et al., 1995; Dudas et al., 2006; Xu et al., 2006; Nakajima et al., 2018). The pattern of Tgf-β3 expression was regulated by Fgf10 in the palatal mesenchyme and the expression of Tgf-β3 in the mid-posterior palatal epithelium was expanded to the oral side in Fgf10−/− mice at stage E13.5 (Alappat et al., 2005). On the other hand, Tgf-β3 also affected the gene expression in the palatal mesenchyme. As aforementioned, Pax9 was a key modulator of a few signaling molecules in palatal epithelium. Its expression was vividly detected in the palatal mesenchyme at E12.5 and became intensified in the medial edge epithelium during fusion (from E13.5 to E14.5) (Sasaki et al., 2007). The expression of Pax9 was significantly downregulated at E13.5 and was completely absent from E14.5 to E15.5 in the palatal region in Tgf-β3−/− mice (Sasaki et al., 2007). Fail in the formation of pseudopods and chondroitin sulfate proteoglycans synthesis might account for the impaired migration of periderm cells in the MEE of the Tgf-β3−/− mice (Taya et al., 1999; Richardson et al., 2017). Tgf-β3 has also been suggested to play a role in regulating EMT via Smad-dependent and Smad-independent signaling pathways to regulate the expression of the EMT-related transcription factors Lef1, Twist, and Snail1, etc., in the epithelial cells in MEE (Yu et al., 2009). In the epithelial cells in the seam, Tgf-β3 also downregulated the expression of p63, which acted directly on the promoters of a variety of genes (Pvrl1, Irf6, Fgfr2, Tcfap2a, Pdgfa, Sfn, Grhl3 and Jag2) that were involved in the determination of the fate of the peridermal cells (Moretti et al., 2010; Thomason et al., 2010). The detailed mechanisms of Tgf-β3 in the regulation of MEE cell fate have been elaborated in excellent reviews by Akira Nakajima et al. (Yu et al., 2009; Nakajima et al., 2018; Hammond et al., 2019; Hammond and Dixon, 2022). Other examples of the epithelial-mesenchymal interaction in regulating palate fusion include mesenchymal Fgf18, which induced the expression of the transcription factor Runx1 in the fusing epithelium from E13.5 to E15.5 (Charoenchaikorn et al., 2009) (Figure 3).

Figure 3. Tgf-β3 is a key player in regulating epithelial fate in MEE. Tgf-β3 is expressed before and after palatal fusion in MEE, where it plays a key role in determining the epithelial cell fate by regulating the expression of a wide range of downstream genes that involved in apoptosis as well as in EMT. Crosstalk between pathways is illustrated using solid lines and dashed lines, indicating direct and indirect regulations, respectively.
7 Summary and perspectivesEpithelial-mesenchymal interactions are one of the fundamental essential developmental events during morphogenesis and organ formation. Mechanisms involved in mediating epithelial-mesenchymal interactions include soluble signaling molecules, direct cell-cell contacts and the components of extracellular matrix. In spite of the sharing molecular mechanisms (i.e., Fgf, hedgehog, Wnt, Tgf-β and Bmps) that govern different organ development, the ectodermal epithelium of facial prominences plays a dominant inductive role during craniofacial development while the instructive function is more likely to be ascribed to the mesenchyme in lung and kidney formation (Ribatti and Santoiemma, 2014). The integrity of facial epithelia that derived from surface ectoderm ensures the spatiotemporal regulation of mesenchymal proliferation and migration as well as subsequent fusion at ventral midline (Lan and Jiang, 2022). The best example came from the development of FEZ, a facial ectoderm domain defined by the reciprocal exclusive expression of Shh and Fgf8 that played morphogenic roles in craniofacial development, as evidenced by numerous conventional knockout and transplantation studies in both chicken and mice as well as by recent quantitative morphometric analysis (Hu et al., 2003; Xu et al., 2015). On the other hand, alternation of mesenchymal signaling actively changed the molecular pattern formation in the ectoderm (Foppiano et al., 2007; Hu and Marcucio, 2012). Compared to the remarkable progress that has been made in resolving the genetic etiology of OFCs by identifying potential causative genes using high throughput technologies, little has been achieved in the understanding of the molecular pathogenesis of these malformations (Sull et al., 2009; Saleem et al., 2019; Yan et al., 2020). Given the active interplay between the facial epithelium and the mesenchyme, effective therapeutic strategies may be developed by intervening in this process.
Author contributionsJL: Writing–original draft. BP: Writing–original draft. WW: Writing–original draft. YZ: Conceptualization, Writing–review and editing, Funding acquisition, Supervision.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was sponsored by the Guangdong Provincial Key R&D Program of Department of Natural Resources of Guangdong Province (#2023B1111050011) and the Natural Science Foundation of Guangdong (#2023A1515010293).
AcknowledgmentsWe thank Miss Shufang Li for her work in drawing the schematic diagram in Figures.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbbreviationsEMI, Epithelial-mesenchymal interaction; OFC, Orofacial cleft; SES, Socioeconomic status; FnP, Frontonasal prominence; MnP, Medial nasal prominence; LnP, Lateral nasal prominence; MxP, Maxillary prominence; MdP, Mandibular prominence; NCC, Neural crest cell; Shh, Sonic hedgehog; Fgf, Fibroblast growth factor; Bmp, Bone morphogenetic protein; Tgf-β, Transforming growth factor-β; Wnt, Wingless-type MMTV integration site; RA, Retinoic acid; EMT, Epithelial-mesenchymal transition; CNCC, Cranial neural crest cell; MEE, Medial edge epithelia; MES, Medial epithelial seam; FEZ, Frontonasal ectodermal zone; Fox, Forkhead box; LOF, Loss-of-function; GOF, Gain-of-function; Osr2, Odd-jump-related 2; Cyp26, Cytochrome P450 subfamily 26; Col, Collagen; ECM, Extracellular matrix; bHLH, basic helix-loop-helix; CLP, Cleft Lip and Palate; RIPK4, Receptor-interacting protein kinase 4.
ReferencesAlappat, S. R., Zhang, Z., Suzuki, K., Zhang, X., Liu, H., Jiang, R., et al. (2005). The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 277, 102–113. doi:10.1016/j.ydbio.2004.09.010
PubMed Abstract | CrossRef Full Text | Google Scholar
Andl, T., Ahn, K., Kairo, A., Chu, E. Y., Wine-Lee, L., Reddy, S. T., et al. (2004). Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 131, 2257–2268. doi:10.1242/dev.01125
PubMed Abstract | CrossRef Full Text | Google Scholar
Ashique, A. M., Fu, K., and Richman, J. M. (2002). Endogenous bone morphogenetic proteins regulate outgrowth and epithelial survival during avian lip fusion. Development 129, 4647–4660. doi:10.1242/dev.129.19.4647
PubMed Abstract | CrossRef Full Text | Google Scholar
Atukorala, A. D. S., and Ratnayake, R. K. (2021). Cellular and molecular mechanisms in the development of a cleft lip and/or cleft palate; insights from zebrafish (Danio rerio). Anatomical Rec. 304, 1650–1660. doi:10.1002/ar.24547
CrossRef Full Text | Google Scholar
Bachler, M., and Neubüser, A. (2001). Expression of members of the Fgf family and their receptors during midfacial development. Mech. Dev. 100, 313–316. doi:10.1016/s0925-4773(00)00518-9
留言 (0)