
記住我
The cell line JHOS2 (Riken BioResource Research Center) was grown in DMEM/F12 (21331020, Gibco, Thermo Fisher Scientific) supplemented with 10% FBS (11573397, Gibco, Thermo Fisher Scientific), 1% UltraGlutamine (BE17-605E/U1, Lonza), 1% MEM nonessential amino acids solution (11140050, Gibco, Thermo Fisher Scientific) and 0.01% Primocin (ant-pm-2, Invivogen). OC cell lines Kuramochi and Ovsaho were obtained from the Japanese Collection of Research Bioresources and grown in RPMI-1640 medium (31870025, Gibco, Thermo Fisher Scientific) supplemented with 10% FBS, 1% UltraGlutamine and 0.01% Primocin. Information on the authentication of cell lines is available in the Nature Portfolio Reporting Summary and their cancer drivers can be found at https://cellmodelpassports.sanger.ac.uk/. The patient material and data were acquired upon informed consent and under institutional ethical review board-approved protocols (no. 56/13/03/03/2014) at the University of Helsinki Central Hospital. The PDCs were cultured as previously described25 but without feeder cells, either in DMEM/F12 (31330038, Gibco, Thermo Fisher Scientific) supplemented with 1× B27 (17504044, Gibco, Thermo Fisher Scientific), 20 ng ml−1 EGF (354052, Corning), 10 ng ml−1 fibroblast growth factor (PHG0023, Gibco, Thermo Fisher Scientific) and 0.02% Primocin or in 75% F12 (21765029, Gibco, Thermo Fisher Scientific) and 25% DMEM (41965039, Gibco, Thermo Fisher Scientific) supplemented with 5% FBS, 0.4 µg L−1 hydrocortisone (H0888, Sigma-Aldrich), 5 µg ml−1 insulin (I0516, Sigma-Aldrich), 8.4 ng ml−1 cholera toxin (C8052, Sigma-Aldrich), 10 ng ml−1 EGF, 24 µg ml−1 adenine (A2786, Sigma-Aldrich), 0.02% Primocin and 10 µM Y-27632 (ALX-270-333, Enzo Life Science). PDCs were grown on Primaria plates (353846, Corning). Cell counting was performed using a TC20 automated cell counter (1450102, Bio-Rad Labotories). During drug treatment, Y-27632 was not used. The PDCs were verified for identical phenocopy with their original tumor samples by next-generation sequencing (data available upon request) and the immunohistochemistry-based expression of the Müllerian marker PAX8, a known marker for the diagnosis of epithelial OCs. All HGSOC PDCs displayed a loss-of-function TP53 mutation and PI3K pathway aberration (PI3KCA gain or amplification).
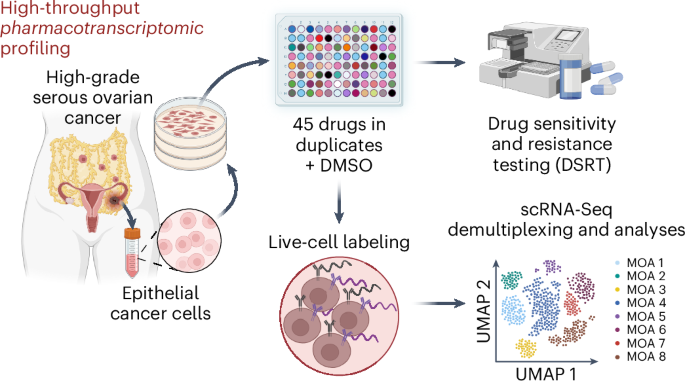
Cell Hashing and scRNA-SeqSample preparation and Cell Hashing labelingThe HGSOC cells from JHOS2 and the two PDCs (PDC2 and PDC3) were harvested according to standard cell culture procedures. After medium removal, the cells were washed with 6 ml of PBS and detached with 1 ml of TrypLE express enzyme (12605010, Gibco, Thermo Fisher Scientific) incubation for 10–15 min at 37 °C and 5.0% CO2 before quenching with 5 ml of warm medium. During incubation, the preprinted drugs in the 96-well plates were dissolved in 25 µl of medium while shaking for 15 min. The cell suspension was then transferred to a 15-ml tube and centrifuged at 300g for 5 min. After removing the supernatant, the pellet was resuspended in 5 ml of warm medium before cell counting, which was performed as described before. The objective was to plate ~13,000 cells per well by adding 75 µl of the cell suspension to each well after dissolving the drugs to obtain a final volume of 100 µl per well of each 96-well plate. Finally, cells were incubated for 24 h at 37 °C and 5.0% CO2 before harvesting for Cell Hashing12. Live-cell labeling was performed as previously described12,55 with pairwise combinations of 20 in-house-produced antibody–oligo conjugates targeting B2M (316302, BioLegend) and CD298 (341712, BioLegend). The full list of drugs used for multiplexed scRNA-Seq, along with their concentrations, identifiers, sources and solvents, is reported in Supplementary Table 1 and the sequences of the 90-nt DNA barcodes conjugated to the antibodies are available in Supplementary Table 3.
Library chemistry, sequencing and raw FASTQ file preprocessingSingle-cell gene expression profiles were studied using the 10x Genomics Chromium single-cell 3′ RNA-Seq platform (10x Genomics). The Chromium single-cell 3′ RNA-Seq run and library preparation were performed using Chromium Next GEM single-cell 3′ gene expression version 3.1 dual-index chemistry with feature barcoding technology (CG000317, 10x Genomics). The sample libraries were sequenced on the Illumina NovaSeq 6000 system (Illumina) using read lengths of 28 bp (read 1), 10 bp (i7 index), 10 bp (i5 index) and 90 bp (read 2). The target minimum coverage was 50,000 reads per cell. Raw FASTQ file preprocessing was performed using Cell Ranger (version 7.1.0, 10x Genomics) pipelines. Specifically, cellranger mkfastq was used to produce FASTQ (raw sequence data) files and cellranger count was used to perform alignment, filtering and unique molecular identifier (UMI) counting. Alignment was performed against the GRCh38 (GENCODE version 32, Ensembl 98)56 assembly of the human genome.
scRNA-Seq data demultiplexing and analysisThe scRNA-Seq data bioinformatic analyses were performed using R (4.2.2)57, primarily using the R package Seurat (4.3.0)58. First, only cells for which both HTO and mRNA data were available were retained. Then, the HTO data were normalized for each of the three batches individually using the function ‘NormalizeData’ with the centered log-ratio transformation. Then, cells were demultiplexed using the function ‘HTODemux’ with default parameters for row-specific and column-specific HTOs, separately. Only cells classified as ‘singlets’ for both the row-specific and the column-specific HTOs were retained, whereas all the negative and doublet cells were discarded. A breakdown of the total number of cells before and after the demultiplexing can be found in Supplementary Table 4. The remaining pool of cells from the HTO classification was subjected to quality controls according to the number of UMIs (nCount_RNA > 2,500 and nCount_RNA < 80,000) and percentage of UMIs from mitochondrial genes (percent.mt < 25 for JHOS2 percent.mt < 30 for PDC2 and percent.mt < 20 for PDC3). After quality control filtering, the batch data were merged for downstream preprocessing. Normalization and variance stabilization of the three batches of scRNA-Seq data were performed using SCTransform v2 (ref. 59), regressing out the percentage of counts accounting for ribosomal and mitochondrial protein-coding genes, as well as the number of genes and UMIs. Cell-cycle heterogeneity was scored after a first round of variance stabilization and normalization with ‘CellCycleScore’, using the cell-cycle phase references provided by Tirosh et al.60 Subsequently, the data were renormalized and stabilized by regressing the cell-cycle scoring. Principal component analysis followed on the single-cell transcriptomic data and the batch effect was corrected with the R package Harmony (version 0.1.1)61. After constructing the shared nearest neighbor network with ‘FindNeighbors’ on the Harmony batch-corrected dimensionality reduction (reduction = ‘harmony’), clustering was performed through use of the Leiden algorithm62 with all the possible resolutions between 0 and 1 and a step size of 0.1; then, ‘clustree’, from the clustree R package (version 0.5.0)63, was used to choose the best resolution (0.3). UMAP embeddings were computed using the ‘RunUMAP’ Seurat function with reduction = ‘harmony’. For all the UMAPs generated, min.dist was set to 0.5, dims was set to 1:30 and the number of neighbors was set to 5. When subsetting for model-specific cells, the data were renormalized and variance-stabilized, without correcting for the batch effect.
Differential expression analyses for the identification of Leiden cluster markers were run using ‘FindAllMarkers’ and adopting the Wilcoxon rank-sum test (test.use = ‘wilcox’). P values were adjusted with Bonferroni correction using all the features in the dataset as default. GSVA64 was conducted using the ‘gsva’ function from the R package oppar (0.99.8)65 against the MSigDB GO:BP C5 collection, as downloaded on March 24, 2023. Gene sets from the C5 collection were filtered to those with at least 10 genes and at most 500 genes, resulting in 834 final gene sets from a total of 7,751. GSVA was calculated using the average expression of the 50 most upregulated and downregulated genes for each of the 13 Leiden clusters as previously computed with ‘FindAllMarkers’. The significance of the scores was calculated by bootstrapping 1,000 times. The top 5% most variable terms were selected by calculating the coefficient of variation of each gene set retained in the analysis. The 3D UMAPs were produced using the R package plotly (version 4.1.10)66 as described by Qadir et al.67
DGE analysis using subsample pseudobulk aggregationEach model and the 46 treatment groups were divided into subsets or subsamples of ten cells68. If the number of cells in a group wasn’t divisible by ten, only subsets with at least five cells were kept. It was ensured that each treatment group had at least two subsets. Consequently, treatment groups with 14 or fewer cells would have been excluded but none were, as even the smallest group had 29 cells. Subsequently, we calculated the pseudobulk expression of each subsample of cells using ‘AggregateExpression’ from Seurat on raw counts. For each of the three models, differential expression analyses were performed using the R packages edgeR (version 3.40.2)69 using the genewise negative binomial generalized linear models with quasilikelihood tests with the functions ‘glmQLFit’ and ‘glmQLFitTest’. Specifically, after filtering out lowly expressed genes (average log2(counts per million) < 1 in the dataset containing the compared subsamples), normalization factors were calculated with ‘calcNormFactors’ and a design matrix was prepared to model the experimental groups. Negative binomial dispersions were estimated with ‘estimateDisp’ and a quasilikelihood negative binomial generalized linear model was fitted to the data. A quasilikelihood F-test with ‘glmQLFitTest’ was performed to compare treatment groups against controls and the genes were extracted with adjusted P values using the Benjamini–Hochberg method. ORA was performed using gprofiler2 (version 0.2.1)70; protein-coding genes that were found significantly upregulated or downregulated (|log2FC| > 0.25, FDR < 0.01) were taken from the DGE analyses results. For these comparisons, the pseudobulk aggregates approach outperformed the canonical one adopted for scRNA-Seq data comparisons, such as the Wilcoxon rank-sum test, in detecting the upregulation of protein-coding genes (Supplementary Fig 3c). Here, for each MOA, the union of the upregulated protein-coding genes across all drugs with an MOA was taken and the same was applied for the downregulated genes. Finally, the two gene sets were independently submitted to ORA against the Reactome28 database, with Homo sapiens as the organism and an FDR threshold of 0.05. The results were filtered for a custom list of cancer-related Reactome pathways derived by merging the pathways identified in the Reactome cancer entry (database identifier: 1500689), pathways significantly enriched in the evolutionary states from Lahtinen et al.30 and Reactome pathways containing the terms ‘FOXO’ or ‘insulin’ (to include terms related to mechanisms of resistance present in HGSOC; Supplementary Table 2). Lastly, if more than 20 Reactome pathways were retained for upregulated or downregulated genes, only the 20 most significant pathways were retained for each of the two sets; for repeated terms, the first and most significant instance was retained.
LINCS perturbagen data analysesA total of 1,436,310 experiment results from the LINCS L1000 chemical perturbations characteristic up and down gene sets were retrieved from the SigCom database71. In these characteristic sets, the top 250 upregulated and downregulated genes for each experiment are presented with experimental conditions. The perturbation experiments were filtered to those that (1) involved a perturbagen classified with an MOA defined as either an ‘AKT inhibitor’ or ‘PI3K inhibitor’; (2) were performed in a cell line from ovary or breast; and (3) had a time point of 24, 48, 72, 96 or 120 h. As a result, 2,254 experiments satisfied these conditions. Count matrices were calculated for all genes that occurred in the top upregulated or downregulated gene sets among the target 2,254 experiments to identify the most commonly perturbed genes by PI3K–AKT inhibition in ovary and breast cells over 24–120 h. Characteristic gene sets for RTK signaling and PI3K–AKT signaling pathways were retrieved from MSigDB72, as part of the C2 curated gene sets, labeled as ‘Reactome signaling by RTKs’ and ‘WP PI3K–AKT signaling pathway’, respectively.
Immunoblotting and immunoprecipitationFor the drug treatment experiments, in the case of the representative cell lines, 250,000 cells per well were seeded to a six-well plate and the cells were treated for 48 h with 100 nM and 500 nM gedatolisib (s2628, Selleck Chemicals), whereas PDCs were treated with 50 nM and 100 nM gedatolisib. An equivalent volume of PBS was added to control (untreated) wells. Cells were lysed with Triton X-based lysis buffer (50 mM Tris-HCl pH 7.5, 10% glycerol, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 (X100-100ML, Sigma-Aldrich) and 50 mM NaF) supplemented with protease and phosphatase inhibitors (phosphatase inhibitor cocktail (two tubes, 100×; B15001) and protease inhibitor cocktail (EDTA-free, 100× in DMSO; B14001), both from Bimake (now Selleck Chemicals)). For immunoprecipitation, cell lysates were incubated overnight at 4 °C with EGFR (4267, Cell Signaling Technology (CST); 1:100) and 1 µg of Ultra-LEAF purified recombinant human IgG1 isotype control (403501, BioLegend) was used as a control. Then, the samples were incubated for 1 h at 4 °C with prewashed protein A agarose beads (9863, CST; 1:4), after which the beads were mixed with 2× Laemmli sample buffer (1610737, Bio-Rad Laboratories) and separated by SDS–PAGE followed by immunoblotting. Briefly, separated proteins from both immunoblotting and immunoprecipitation were transferred to 0.45-µm nitrocellulose membranes and the following primary antibodies were used (all 1:1,000 dilution): anti-β-tubulin (86298, CST), anti-AKT (2920, CST), anti-pAKT (S473) (4060, CST), anti-CAV1 (3267, CST), anti-EGFR (4267, CST), anti-ERK1–ERK2 (4696, CST) and anti-pERK1–pERK2 (9101, CST). Secondary antibodies were IRDye 800CW donkey anti-mouse IgG and IRDye 680RD donkey anti-rabbit IgG (926-32212 and 926-68073, LI-COR) or AzureSpectra fluorescent secondary antibodies goat anti-mouse 550 (AC2159, Azure Biosystems). The membranes were scanned with the Odyssey Fc imaging system (LI-COR) or Azure 600 (Azure Biosystems). Image analysis was performed using Image Studio Lite (LI-COR).
Immunofluorescence stainingA total of 1,000 JHOS2 and 5,000 Kuramochi cells were seeded on 12-mm glass coverslips on 24-well plates for 24 h. In the case of PDCs, 30,000 PDC1, PDC2 and PDC3 cells were seeded. Cells were treated with 500 nM afuresertib (FIMM136448, FIMM), AZD-8186 (A-1610, Active Biochem), gedatolisib, idelalisib (FIMM003750, FIMM), pictilisib (HY-50094, MedChemExpress) and sapanisertib (FIMM109442, FIMM) for 48 h. Then, the cells were fixed for 10 min with 4% paraformaldehyde pH 7.4 (158127, Sigma-Aldrich) followed by permeabilizing the cells with 0.1% Triton X-100 solution in PBS for 10 min. After blocking the cells with 1% BSA (P6154, BioWest) for 1 h at room temperature, cells were incubated overnight with anti-EGFR and anti-CAV1 antibodies (as used for immunoblotting; 1:500) at 4 °C. As a secondary antibody, donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody, AlexaFluor Plus 594 (A32754, Thermo Fisher Scientific; 1:1,000) was used, whereas DAPI (D9542, Sigma-Aldrich) was used for nuclear staining. The glass coverslips in the 24-well plate were picked and mounted on glass slides with Immu-Mount (9990402, Fisher Scientific). Images were captured with LSM700 Confocal Microscope (Carl Zeiss) and inspected with ZEN Blue digital imaging for light microscopy (version 3.5.093, Carl Zeiss).
DSRT and small-scale drug combination testingDSRT and the calculation of the DSSs were performed as described previously26,73. In short, 1,000–1,500 cells were added to wells of 384-well plates with drugs preplated with five concentrations over a 10,000-fold concentration range. After a 3-day incubation at 37 °C and 5.0% CO2, cell viability was measured using the CellTiter-Glo 2.0 assay (G9242, Promega), luminescence was read with a PHERAstar FS (BMG Labtech) and the outcoming data were analyzed using Breeze74 (https://breeze.fimm.fi/). For small-scale drug combination testing, 1,200 cells were seeded to a 384-well plate in 25 µl of medium, including the drugs or drug combinations in desired concentrations. Gefitinib was used at a concentration of 1 µM for PDCs and 10 µM for cell lines, while other drug (gedatolisib, afuresertib, AZD-8186, idelalisib, pictilisib and sapanisertib) concentrations varied depending on their properties; concentrations are reported in the relative figure legends. Five technical replicates were used for each sample. After 72 h of incubation, cell viability was measured as described above with CellTiter-Glo 2.0 and luminescence was read with PHERAstar FS or Spark multimode microplate reader (Tecan Group). The CellTiter-Glo data analysis was conducted using GraphPad Prism software (version 9.3.1) to obtain comprehensive results and statistical analysis. The s.d. was chosen over the s.e.m. to provide a measure of the variability within the data points rather than the precision of the estimated mean.
Image-based small-scale drug testing of PDOsEarly-passage PDCs were embedded in 25 µl of 66% VitroGel ORGANOID-4 (VHM04-4, TheWell Bioscience) according to the manufacturer’s recommendations on a 384-well plate (ultralow attachment, Corning), with 5,000 cells per well. The plate was incubated at 37 °C for 15 min followed by the addition of 25 µl of top medium. The top medium was refreshed every 4 days. Organoids were allowed to form over 14 days before treatment using the following drugs: 50 nM gedatolisib and 500 nM gefitinib as monotreatment or in combination, diluted in medium. As a control, an equivalent volume of DMSO was added and three replicates were used. After 12 days of drug exposure, cells were live-stained with 75 nM TMRE (Abcam), and z-stacked images were taken using Leica THUNDER Imager live cell and 3D assay (Leica). The images were analyzed using CellProfiler (version 4.2.1)75 and the standard pipeline (including modules ‘IdentifyPrimaryObjects’ and ‘MeasureImageAreaOccupied’) was used to analyze the area of the image covered with the TMRE signal to quantify the viability of organoids in each condition. Results were standardized to the control.
siRNA-mediated silencing and overexpression of CAV1 and EGFRTransient silencing of CAV1 and/or EGFR was achieved by plating JHOS2 and Kuramochi cells in six-well plates with 200,000 cells per well for immunoblotting analyses and in 96-well plates with 8,000 cells per well for cell viability assays. In both settings, the cells were allowed to adhere to the plates for 24 h, after which the transfections were performed. For the silencing of CAV1 (L-003467-00-0005, Dharmacon Reagents), EGFR (L-003114-00-0005, Dharmacon Reagents) or the control (D-001810-10-05, Dharmacon Reagents), an ON-TARGETplus siRNA smart pool of four siRNAs per target was used at the final concentration of 12.5 nM using DharmaFECT 1 transfection reagent (T-2001-02, Dharmacon Reagents). For plasmid transfections, CAV1 was cloned with a Flag tag into the pCDNA3.1 expression plasmid and 2 µg of purified DNA per well in six-well plates or 100 ng of purified DNA per well in 96-well plates was transfected using FuGENE HD transfection agent (E2311, Promega) as per the manufacturer’s instructions. Equal amounts of pSG5 plasmid (216201, Agilent Technologies) were used in transfections as a control. Drug treatment was performed 24 h after transfections at the concentrations indicated in the figure legends. Then, 48 h after the drug introduction, the cells were harvested by whole-cell lysis and used for immunoblotting or subjected to cell viability assay as described above.
Colony-forming assayDepending on the growth rate of the cell model, 5,000–20,000 cells per well were plated in 12-well plates. Then, 24 h later, PDCs were treated with either gedatolisib (PDC2, 50 nM; PDC3, 25 nM) or gefitinib (500 nM) as monotherapy or in combination; the representative cell lines were treated with gedatolisib (100 nM) and gefitinib (10 µM) alone or in combination. The growth medium and drugs were refreshed every third day. Treatment continued for 8–12 days until the untreated cells were confluent. Subsequently, the cells were fixed with a solution of methanol and acetic acid (7:1) and stained with 0.5% crystal violet (C3886, Sigma-Aldrich). The images of the plates were taken using imageRUNNER ADVANCE C2225i (Canon) for qualitative analysis followed by eluting the bound crystal violet stain with 100% methanol (10653963, Fisher Chemical). The absorbance of the elute was measured at 620 nm using the Spark multimode microplate reader (Tecan Group).
Bulk RNA-Seq library chemistry, sequencing and raw FASTQ files preprocessingTotal RNA was isolated from the PDCs using the RNeasy kit (Qiagen). The quantity and quality of the RNA samples were assessed by Qubit (Thermo Fisher Scientific) and Bioanalyzer (Agilent Technologies). RNA with an RNA integrity number > 8 was used for subsequent analysis. Libraries were multiplexed and paired-end sequencing was performed with the Illumina HiSeq system (Illumina). Resulting FASTQ files were aligned to the GENCODE version 36 (GRCh38)76 assembly of the human genome with the STAR RNA-Seq aligner (version 2.7.10b)77. STAR run parameters were defined as follows: ‘--twopassMode Basic’, ‘--alignSJoverhangMin 8’, ‘--alignSJDBoverhangMin 1’, ‘--outFilterMismatchNoverLmax 0.1’, ‘--alignIntronMin 20’, ‘--alignIntronMax 1000000’, ‘--outFilterScoreMinOverLread 0.33’, ‘--outFilterMatchNminOverLread 0.33’, ‘--limitSjdbInsertNsj 1200000’, ‘--outSAMstrandField intronMotif’, ‘--chimSegmentMin 15’, ‘--chimJunctionOverhangMin 15’ and ‘--chimMainSegmentMultNmax 1’; all other parameters were default.
Cell type deconvolution of OC from TCGASample and clinical data and raw counts for 429 bulk RNA-Seq analyses were downloaded from TCGA OC dataset (TCGA-OV)29,78. Unstranded counts were extracted from all experiments and combined. scRNA-Seq count data were downloaded for 43,945 ovary cells with associated cell metadata annotations, including cell type (endothelium, epithelium, erythrocyte, fibroblast, lymphocyte, myeloid, plasma or undefined) and sample location (normal, tumor or tumor-adjacent)79,80. TCGA-OV bulk RNA-Seq count data were then deconvoluted using the R package BayesPrism (version 2.0)81 and the scRNA-Seq count and annotation data were used to identify overall cell type composition and gene expression by cell type for all 429 samples. Deconvoluted raw counts were subsequently normalized with the ‘vst’ function of the R package DESeq2 (version 1.40)82 and then normalized to transcripts per million (TPM) based on GENCODE version 36 gene exon annotations.
CAV1 transcript analysisData for 11,045 ChIP-Seq experiments that were uniformly processed with MACS2 (ref. 83) were retrieved as BED files from the Gene Transcription Regulation Database84. ChIP-Seq peak intervals were mapped to the promoter of the CAV1 gene (Ensembl release 109 annotation) for MYC (147 experiments), MYCN (47 experiments), EGR1 (26 experiments) and TP53 (159 experiments) with CAV1 transcripts plotted using the R package genemodel (version 1.10)85. Transcript-specific expression values as TPM were downloaded for bulk RNA-Seq experiments cataloged in the Genotype-Tissue Expression project (version 8)86 to compare the expression values of CAV1 transcripts in 195 normal ovary samples with those of PDC1, PDC2 and PDC3. Transcript-specific expression values for PDC1, PDC2 and PDC3 were quantified using the ‘quant’ function of the kallisto program (version 0.46.1)87 with Ensembl genomes version 96 as the index.
Statistical informationThe statistical techniques used for scRNA-Seq and RNA-Seq analyses were elucidated in the preceding sections. Pertinent details concerning the statistical analyses were incorporated within the figure legends or described in the Methods, wherever applicable.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)