
記住我
All zebrafish experiments were performed in accordance with the UK Animals (Scientific Procedures) Act with appropriate Home Office Project and Personal animal licenses and with University of Cambridge Animal Welfare and Ethical Review Body approval. Studies were performed in accordance with PREPARE and ARRIVE guidelines. Zebrafish were maintained on a 14-h light/10-h dark cycle under standard conditions55.
All transgenic zebrafish lines used in this study are registered on the Zebrafish Information Network (ZFIN) database and are listed as follows. The retinal models used to assess photoreceptor degeneration were the transgenic line Tg(rho:EGFP-Hsa.MAPT)cu7, referred here to as rho:eGFP–tau-WT and expresses human wild-type tau in rod photoreceptors, and the rho:eGFP line Tg2(rho:EGFP)cu3, which expresses GFP in rod photoreceptors; both were first described by Moreau and colleagues14. The transgenic line expressing mutant tau-P301L is assigned Tg(rho:EGFP-Hsa.MAPT_P301L)cu12 on the ZFIN database and is referred to as rho:eGFP–tau-P301L here. This line was generated in-house as previously described13. The Gal4 driver line Tg3(Xla.Eef1a1:GAL4-VP16)cu11 was generated in-house and is referred to as EIf1a:Gal414, and the pan-neuronal Gal4VP16 driver line (referred to as PanN:Gal4) was a kind gift from H. Baier, Max Planck Institute for Biological Intelligence, Munich, Germany (identified as s1101tEt in the original publication56). Transgenic zebrafish lines expressing Dendra2 fused to either wild-type tau, Tg(UAS:Dendra2-Hsa.MAPT,myl7:EGFP)cu9 referred to as Dendra–tau-WT, or mutant tau-A152T, Tg(UAS:Dendra2-Hsa.MAPT_A152T,myl7:EGFP)cu10 referred to as Dendra–tau-A152T, were made in-house as previously described17. The transgenic line expressing Dendra-tagged tau-P301L was generated in-house as previously described for fish expressing Dendra-tagged tau-A152T17. The transgenic line is assigned Tg(UAS:Dendra2-Hsa.MAPT_P301L,myl7:EGFP)cu61 on the ZFIN database and is referred to as Dendra–tau-P301L.
The atg7+/− (atg7sa14768) mutant fish line was obtained from the Zebrafish Mutation Project57 and was used for generating autophagy-null fish, as previously described4.
Experimental crossesEmbryos from natural spawnings were collected in embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM Mg2SO4 and 5 mM HEPES, pH 7.2), staged according to established criteria58 and reared at 28.5 °C in the dark in an incubator. Petri dishes containing embryos were cleaned daily, and embryo medium was replenished when needed. Eggs collected from a single breeding tank are termed a clutch.
Crosses of the homozygous rho:eGFP–tau-WT line to Tupfel Longfin (TL) wild-type zebrafish or incrosses of the transgenic line were used to generate heterozygous or homozygous offspring for primary screening and validation experiments, respectively. Offspring from crosses of the rho:eGFP–tau-P301L line to wild-type TL zebrafish were used for further investigation of methocarbamol. The embryos from these rho promoter lines were treated with 0.003% phenylthiourea (PTU) in embryo medium from 1 d.p.f. to block pigment formation and screened for eGFP fluorescence in the eyes at 4 d.p.f. on a Leica M205 FA fluorescence microscope. Fish were then washed in embryo medium to remove PTU. In clutches from crosses of homozygous rho:eGFP–tau-WT fish, all offspring express the transgene, and, hence, no PTU was required.
Crosses of the Dendra–tau lines to PanN:Gal4 resulted in fish with pan-neuronal expression of Dendra–tau transgenes throughout the CNS. These were used to confirm drug activity and to evaluate the effects of pharmacological and genetic modulation of CAs on the phenotypic abnormalities and biochemical defects previously described in these lines17. Crosses of the Dendra–tau lines with the Eif1a:Gal4 driver line resulted in mosaic expression of Dendra–tau in all tissues and were used to measure tau clearance kinetics in individual neurons in the spinal cord18.
Crosses of rho:eGFP–tau-WT fish to atg7+/− fish were used to generate rho:eGFP–tau-WT; atg7+/− fish. Crosses of rho:eGFP–tau-WT; atg7+/− fish to atg7+/− fish resulted in a proportion of rho:eGFP–tau-WT; atg7−/− offspring unable to perform autophagic degradation.
Primary screen Treatments with compound libraryA panel of 1,437 drugs from the JHCCL15 were tested in the zebrafish model rho:eGFP–tau-WT to identify new potential tau toxicity modulators.
The compound library was provided in a 96-well-plate format. Compounds were solubilized in either DMSO or water at a final concentration of 100 mM and stored at −80 °C. On the day before the start of drug treatment, a daughter plate was prepared at a concentration of 10 mM in the appropriate solvent. The daughter plates contained sufficient quantities to complete the 5-day treatment and were stored in the dark at room temperature in a desiccator chamber. Working dilutions were identified by codes without any correspondence to its name or clinical indication.
For the primary screen, offspring from crosses of the homozygous rho:eGFP–tau-WT line to wild-type TL zebrafish were collected, cleaned, counted and kept in the incubator until 6 d.p.f. Larvae from a single clutch were then arrayed in a 24-well plate with ten larvae per well, with each well containing 1 ml of embryo medium. One microliter of compound (10 mM) was added to each well, resulting in a final concentration of 10 μM in the well. For each clutch, one plate included vehicle control wells (water and 0.1% DMSO) and one well treated with 10 μM clonidine as a positive control. Larvae were treated from 6 to 10 d.p.f. with 0.5 ml of embryo medium and 0.5 µl of compound replenished daily. Fish health and motility were monitored throughout the treatment period, and signs of toxicity or swimming impairment were noted; larvae were culled immediately if toxicity was observed. At 10 d.p.f., fish were culled by an overdose of 1,3-amino-benzoic acid ethylester (MS222), and samples were collected, dried and stored at −80 °C for subsequent western blotting.
For validation of the results from the primary screen, new commercial stocks were purchased for the selected compounds and tested over a range of concentrations in homozygous larvae from incrosses of homozygous rho:eGFP–tau-WT fish because these presented with more severe retinal degeneration. Four replicates were included for these analyses.
Western blotting analysis of rod photoreceptor degenerationQuantification of rod photoreceptors in fish samples from the primary screen and validation experiments was performed by measuring the amount of endogenous rod-specific rhodopsin protein (zpr-3, ZIRC; 1:250; 36 kDa) compared with the cone-specific protein arrestin-3 (zpr-1, ZIRC; 1:500; 40 kDa) by western blotting, as previously described14.
Imaging of rod photoreceptor degenerationQuantification of rod degeneration in rho:eGFP–tau lines was performed on images from 10-μm cryosections across the central retina of larvae fixed at 10 d.p.f., as previously described4. A minimum of 24 eyes were quantified for these experiments, and data are represented as photoreceptor area in pixels.
eGFP–tau expression in rho:eGFP–tau-WT fisheGFP–tau expression in drug-treated rho:eGFP–tau-WT fish was evaluated using RNA isolated from pools of ten larvae at 10 d.p.f. extracted using an RNeasy Plus Mini kit (Qiagen) following the manufacturer’s instructions. Fish treated with 0.1% DMSO or 3 μM methocarbamol were compared with untreated siblings. A total of 50 ng of RNA from each condition was used in One-Step qPCR combining cDNA synthesis and real-time PCR. We used customized TaqMan gene-specific primers for EGFP and GAPDH as loading controls and followed the manufacturer’s instructions (Invitrogen; GAPDH TaqMan made-to-order gene expression code 4351372 Dr03436845_g1 and EGFP TaqMan made-to-order code 6302625 from Applied Biosystems). Three independent clutches were evaluated in triplicate and analyzed on a StepOne Plus Real Time PCR System using StepOne Software V.2.1 (Applied Biosystems, Life Technologies). Relative gene expression was normalized to that of GAPDH and calculated using the \(2^_}\) method.
Immunostaining to detect phosphorylated tau in the retinal modelCryosections across the central retina of rho:eGFP–tau-WT and rho:eGFP–tau-P301L fish at 10 d.p.f. were used to investigate the effects of 1 µM methazolamide on phosphorylated tau. Antibody staining for the tau marker AT8 (Ser 202–Thr 205) was performed as previously described14. Images of eGFP signal (green) and AT8-specific signal (red) from a minimum of 26 eyes per group were acquired with a Zeiss Axio Zoom.V16 microscope with a QImaging Retiga 2000 R digital camera and QCapture Pro 7.0 software. Images were merged using Fiji software (ImageJ 1.54f) to show the proportion of rods with AT8+ staining. The number of green (GFP+) and red (anti-AT8+) rods was quantified manually in both images and is represented as the percentage of rods showing AT8 staining (see representative images in Extended Data Fig. 3e(ii)).
Phenotypic assessment in Dendra–tau modelsEmbryos with pan-neuronal expression of Dendra–tau-A152T develop abnormal morphological phenotypes, as previously described17. Expression of both tau-A152T and tau-P301L transgenes results in a proportion of fish with bent bodies in ‘S’ and ‘C’ shapes. Phenotypes were scored as being normal or having mild, moderate or severe morphological defects by using a dissecting microscope. The quantification of the number of fish in each category was performed at 3 d.p.f. This analysis has been used previously to assess the effect of pharmacological and genetic modulators of tau toxicity4,13,17.
Phenotypic assessment following drug treatment was performed by adding compounds at the indicated concentrations to the embryo medium at 1 d.p.f. Drug concentration was maintained in embryo medium by replenishing the drug and medium daily until 3 d.p.f., when phenotypes were scored blind to limit biased interpretation of the treatment. Drugs were tested at the following concentrations; methocarbamol at 3, 10 and 30 μM and methazolamide, acetazolamide and tioxolone at 0.1, 0.3, 1, 3, 10, 30 and 100 μM. Fish were monitored daily, and any adverse effects were noted.
Western blottingSamples for western blotting were prepared and processed as previously described17. The primary antibodies used were Tau5 mouse anti-tau (1:1,000; 80579, Abcam), mouse anti-tubulin (1:5,000; T6199, Sigma-Aldrich), mouse anti-PHF1 (tau phosphorylated at Ser 396–Ser 404 used at 1:100; a kind gift from P. Davies, Albert Einstein College of Medicine of Yeshiva University), rabbit anti-LC3 (1:1,000; NB100-2220, Novus Biologicals), rabbit anti-CA IX (H-11) for Ca9 (1:500; sc-365900, Santa Cruz), rabbit anti-CA IV (G-11) for Ca4 (1:500; sc-74527, Santa Cruz) and anti-β-actin (1:1,000; A5316, Sigma-Aldrich). The secondary antibodies used were goat anti-mouse horseradish peroxidase (HRP; 1:5,000; P044701-2, Agilent) and goat anti-rabbit HRP (1:5,000; P044801-2, Agilent).
Immunoreactive bands were detected by the addition of enhanced chemiluminescence (ECL) substrate (GE Healthcare Bioscience) using Hyperfilm ECL (Amersham) and a Fujifilm FPM-100A developer, except for images of sarkosyl-soluble and sarkosyl-insoluble tau in Fig. 1f,g, which were acquired using an LI-COR Odyssey Fc and Image Studio software (version 5.2). Images were digitized, and densitometry of the bands was quantified using ImageJ (Fiji) software.
Fractionation of soluble and insoluble tauPools of 50 embryos with pan-neuronal expression of Dendra–tau-A152T were collected at 6 d.p.f. after being treated with 0.1% DMSO or 3 μM methocarbamol from 1 d.p.f. and frozen at −80 °C. The extraction of soluble and sarkosyl-insoluble tau fractions was performed using the standard protocol1 with some modifications for fish samples, as previously described17.
Tissue-specific CA gene expressionEye-specific expression of different zebrafish CAs was evaluated by extracting RNA using a Norgen Single Cell RNA Purification kit (51800) according to the manufacturer’s protocols from ten whole bodies or pools of 40 eyes from rho:eGFP–tau-WT fish at 9 d.p.f. cDNA was synthesized from 1 μg of RNA using an AB Biosynthesis cDNA kit (4368814, Applied Biosystems) according to manufacturer’s instructions. cDNA was then used to perform PCR with a temperature gradient (ranking from 55 to 65 °C) for the annealing step to determine the optimal annealing conditions for each primer pair for each CA isoform. Primers were designed for the genes encoding the most abundant cytosolic isoforms Ca2a and Cahz; membrane-bound isoforms Ca4a, Ca4b, Ca4c, Ca9 and Ca14 and the mitochondrial ortholog Ca5 (see Supplementary Table 4). PCR products were then resolved on agarose gels to visualize bands at the specific expected sizes.
Expression of CAs in Dendra–tau+ neurons from fish with pan-neuronal expression was investigated by isolation of Dendra+ cells by FACS. Fish were culled at 3 d.p.f. by overdose of anesthesia and collected into tubes (50 fish per tube). After yolk sac removal by pipetting up and down several times in 500 μl of deyolking solution (55 mM NaCl, 1.8 mM KCl and 1.25 mM NaHCO3), tissue was disaggregated in 900 μl of liberase solution (0.075 mg ml–1 Roche Liberase Research Grade in DEPC-PBS) by mechanical disruption, pipetting up and down every 2–3 min for no longer than 20 min to avoid inducing cell death at room temperature. Then, 100 μl of fetal bovine serum (FBS; F7524, Sigma-Aldrich) was added, and tubes were placed on ice to stop the reaction. The dissociated cell suspension was then filtered through a 30-μm cell strainer (Filter Cell Trics 30 μm, Sysmex, 04-0042326) into a 15-ml tube on ice. The filter was then washed with 10 ml of cold DEPC-PBS, and the eluate was collected in the same tube. Cells were pelleted by centrifugation at 600g for 3 min at 4 °C and resuspended in 500 μl of cold DEPC-PBS by gently pipetting. Dendra+ neurons were then sorted using a FAC-SORTER MoFlo Astrios.
RNA was extracted from a minimum of 150,000 cells using a Norgen Single Cell RNA Purification kit (51800) and reverse transcribed to cDNA using an AB Biosynthesis cDNA kit according to manufacturer’s protocols. Analysis of CA expression by PCR was performed as described above for whole bodies and eyes.
CRISPR injectionsA total of four CRISPR guide RNAs were custom designed for each eye and/or CNS-expressed CA isoform by Horizon Discovery (Dharmacon; listed in Supplementary Table 5). A mixture of all four CRISPR guide RNAs was used to maximize the silencing of each gene59. One hundred nanograms of each CRISPR guide RNA was mixed with 800 ng of transactivating CRISPR RNA (tracrRNA; Dharmacon Edit-R CRISPR–Cas9 synthetic tracrRNA; U002005, Dharmacon) in a total volume of 9.6 µl and incubated at room temperature for 10 min. After incubation, 2.64 µl of 2 M KCl was added, and the final volume was aliquoted into 1.5-µl aliquots and stored at −80 °C until use.
On the day of injection, an aliquot of CRISPR + tracrRNA was thawed on ice and mixed with 5 µg of Cas9 Nuclease Protein NLS (Horizon Discovery, CAS12206). After 5 min of incubation at 37 °C, 0.2 µl of phenol red was added to the solution.
Embryos from homozygous rho:eGFP–tau-WT fish crossed with TL wild-type fish or Dendra–tau-A152T fish crossed with the PanN:Gal4 line were collected immediately after spawning and injected into the yolk with 4.28 nl of CRISPR–Cas9 solution while at the one-cell stage. Developing embryos were thoroughly cleaned a few hours after injection and monitored daily.
Knockdown efficiency by CRISPR injectionThe efficiency of CA-targeting CRISPR guide RNAs was evaluated by qPCR. CRISPR injections were performed in five independent clutches of wild-type embryos as described above. Uninjected fish from each clutch were collected for comparison. At 4 d.p.f., pools of ten fish from each group were collected, and RNA was extracted using an RNeasy Plus Mini kit (Qiagen) according to manufacturer’s instructions. A total of 1 μg of RNA from each condition was used to generate cDNA using an AB Biosynthesis cDNA kit (4368814, Applied Biosystems) following the manufacturer’s instructions. cDNA was then mixed with SYBR Green qPCR Master Mix (Thermo Fisher Scientific, 330501) and the relevant CA primer pairs targeted to the C-terminal exons (listed in Supplementary Table 6). qPCR was performed in a Roche LightCycler 480 machine using LightCycler 480 Software (V1.5.1.62). Gene expression values were normalized to rbs11 as a loading control and calculated using the \(^_}\) method.
Analysis of Dendra–tau clearance kineticsThe clearance rate of tau following drug treatment was determined as previously described17,18. Offspring from Dendra–tau-WT, Dendra–tau-A152T or Dendra–tau-P301L fish crossed to the EIF1a:Gal4 line have mosaic expression of the transgenes and were used to measure Dendra-tagged tau in individual neurons in the spinal cord. To investigate changes in Dendra–tau clearance kinetics after CA inhibition, 3 μM methocarbamol or 1 μM methazolamide was added to embryo medium after photoconversion, whereas the control group was treated with 0.1% DMSO. To study the influence of lysosomal and proteasomal degradation in Dendra–tau clearance, fish were treated with 3 μM methocarbamol in the presence or absence of 10 mM NH4Cl or 100 mM MG132, respectively. Drugs were replenished every 12 h.
The effect of genetic inhibition of CAs on the Dendra–tau-A152T clearance rate was evaluated in fish injected with CRISPR guide RNAs targeting Ca4a (ca4a) at the one-cell stage in the presence or absence of 1 μM methazolamide added immediately after photoconversion and compared with their respective uninjected control siblings.
Proteasome activity assayThe chymotrypsin-like activity of the proteasome was evaluated in fish homogenates as previously described17 with the following modifications. Fish with pan-neuronal expression of Dendra–tau-WT were treated with 3 μM methocarbamol or 0.1% DMSO for 24 h from 1 to 2 d.p.f. Pools of ten fish from three independent clutches were processed as previously described, and 40 μg of protein from each sample was used for the chymotrypsin-like activity assay measuring fluorescence intensity (excitation 355 nm; emission 460 nm) every 5 min over 3 h at 28 °C in a FLUORstar Omega fluorometer (BMG Labtech) with Omega software (V6.20). All samples were measured in triplicate.
‘In vitro’ analysis of the effect of methocarbamol on proteasomal activity was performed on homogenates from fish with pan-neuronal expression of Dendra–tau-A152T and nonexpressing siblings at 2 d.p.f. following the same protocol described above. Once 40 μg of protein was plated and before the addition of 100 mM Suc-LLVY-AMC substrate, methocarbamol was added to the well at final concentrations of 1, 3 or 30 μM. Changes in fluorescence intensity were monitored as described above.
Cell-based experimentsCell cultures were maintained under normoxic conditions (5% CO2) at 37 °C with regular PCR evaluation to ensure that all cell lines were free from mycoplasma contamination.
Analysis of tau clearance in cell cultureInducible SH-Tau cells (eGFP–tau-P301L)60 were maintained in DMEM/F12 medium (D6421, Sigma-Aldrich) supplemented with 10% FBS (F7524, Sigma-Aldrich), 2× nonessential amino acids (M7145, Sigma-Aldrich), 100 U ml–1 penicillin–streptomycin (P0781, Sigma-Aldrich) and 2 mM l-glutamine (G7513, Sigma-Aldrich). Cells were incubated in a humidified incubator at 37 °C and 5% CO2. For clearance assays, with or without transfection of siRNAs targeting VAMP7 and ARL8B, cells were cultured on poly-d-lysine-coated (50 mg ml–1; P6407, Sigma-Aldrich) 96-well, black-walled, flat-bottom μClear plates (655986, Greiner Bio-One) up to a confluency of 70–80%.
siRNA transfectionFor clearance assays with siRNA transfection, cells were first transfected with VAMP7 and ARL8B siRNA, followed by drug treatment. Cells were reverse transfected with specific individual siRNAs targeting VAMP7 and ARL8B (sequence details are provided in ‘Cell transfection with siRNA’) in 96-well plates (as described above) with siRNA (30 nM final concentration per well) in growth medium without antibiotics. After 4–6 h, the transfection medium was replaced with normal growth medium. After 24 h, cells were split 1:3 using trypsin (T3924, Sigma-Aldrich). The following day, cells were forward transfected with 30 nM siRNA as described before. After 24 h, cells were split 1:2 in 96-well plates and incubated at 37 °C for another 24 h.
Drug treatment (with and without siRNA transfection)After 24 h, cells were washed once with serum-free medium (DMEM/F12 without supplements). After washing, cells were loaded with 1.2 µM Cell Tracker Red CMTPX (C34552, Molecular Probes, Invitrogen) in serum-free medium (DMEM/F12 without supplements) for 30 min at 37 °C. Cell Tracker Red was used as an internal control for the number of cells in the well. After incubation, the cells were washed once with serum-free medium (DMEM/F12 without supplements). Cells were reverse treated (that is, tau expression was switched on and drug treatment was applied at the same time) by the addition of doxycycline (0.2 μg ml–1) in combination with methocarbamol at concentrations of 1, 3, 10 or 30 μM or methazolamide at concentrations of 0.1, 1, 3, 10 or 30 μM. Rapamycin (0.4 μM), torin (1 µM) and trehalose (100 mM) were used as positive controls, and cells treated with or without DMSO were vehicle and negative controls, respectively. After 6 h, the plates were scanned by a Tecan Spark Trading microplate reader (SPARK multimode microplate reader with software SPARKCONTROL Method Editor version 3.0) for t = 0 for red excitation/emission spectra (577 nm/602 nm) and green excitation/emission spectra (484 nm/535 nm). Plates were subsequently scanned daily for 4 days. Treatments were refreshed after 48 h, and tau expression was switched off by removing doxycycline from the treatment at 48 h. Three independent experiments and four independent experiments were performed for methocarbamol and methazolamide, respectively. Data for tau levels were analyzed as green fluorescence intensity (for eGFP–tau-P301L) corrected for red fluorescence (for Cell Tracker CMTPX, an internal control for the total number of cells) for each plate (replicate). Corrected values were normalized to the mean of three control (DMSO-treated) plates (three replicates) of each independent experiment at each time point. Data were pooled as means of each independent experiment at each time point. Statistical analysis was performed using two-way ANOVAs followed by Dunnett’s multiple comparisons. All analyses were performed in Prism.
Analysis of tau secretion Immunoprecipitation of GFP–tau-P301LTau secretion was analyzed using the inducible SH-Tau cell line expressing GFP–tau-P301L. Cells from the monoclonal SH-SY5Y cell line expressing GFP–tau-P301L (from Y.-F. Liao’s group Institute of Cellular and Organismic Biology, Taipei, Taiwan) were cultured in DMEM/F12 (D6421, Sigma-Aldrich) supplemented with 10% FBS (F7524, Sigma-Aldrich), 2 mM l-glutamine (G7513, Sigma-Aldrich) and 100 U ml–1 penicillin–streptomycin (P0781, Sigma-Aldrich) for 3 days on six-well plates to 50% confluency. Cells were then treated with 0.2 µg ml–1 doxycycline for 24 h to induce GFP–tau-P301L expression. After doxycycline induction, cells were washed three times in PBS and incubated in complete medium containing doxycycline (0.2 µg ml–1) in the presence of 0.1% DMSO, 100 nm bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for 3, 6, 9 or 12 h. At each indicated time (one individual well per time point), 500 μl of culture medium was collected, and cells were lysed in Laemmli sample buffer and boiled for 10 min at 100 °C.
Tau levels in the cell medium were analyzed by immunoprecipitation of GFP–tau-P301L using GFP-Trap magnetic beads (gtma-20, ChromoTek). Culture medium (500 μl) was incubated with 25 μl of prewashed GFP-Trap beads for 1 h at 4 °C on a rotating surface. GFP beads were washed three times with PBS, resuspended with Laemmli sample buffer and boiled for 10 min at 100 °C. Levels of GFP–tau-P301L in the cell lysates were analyzed by western blotting, a LI-COR Odyssey CLX and Image Studio software for western blot imaging. Cell lysates and GFP-Trap immunoprecipitated tau from cell medium were resolved by SDS–PAGE and transferred onto PVDF membranes. PVDF membranes were blocked with 4% skim milk in phosphate-buffered saline with 0.1% Tween 20 (PBST) for 1 h and incubated overnight at 4 °C with the following primary antibodies diluted in 4% skim milk in PBST: rabbit polyclonal anti-GFP (1:1,000; ab6556, Abcam) and rabbit anti-β-actin (1:1,000; A2066, Sigma-Aldrich). After several washes in PBST, membranes were incubated for 90 min at room temperature with an anti-rabbit HRP-linked antibody (1:5,000; 7074, Cell Signaling Technology) diluted in 4% skim milk in PBST. GFP–tau-P301L and β-actin bands were detected using the ECL enhanced chemiluminescence detection kit from GE Healthcare (RPN2106). Experiments were performed in triplicate.
Split luciferase complementation assayTau secretion was analyzed by a split luciferase complementation assay in cells expressing HiBit-tagged tau. To generate an SH-SY5Y cell line stably expressing HiBit-tagged tau, we transfected SH-SY5Y cells with pcDNA3.1-GFP-2A-HiBit-Tau (810390DE, Thermo Fisher Scientific) using TransIT-2020 transfection reagent (5405, Mirus), following the manufacturer’s recommendations. After 2 days, transfected cells were selected with 600 μg ml–1 G-418 (4727878001, Sigma-Aldrich) for 1 week, after which GFP+ cells were isolated by FACS, and single cells were collected in 96-well plates. After expansion into six-well format, GFP and HiBit tau expression were assessed from clonal lines by flow cytometry and immunofluorescence microscopy analysis.
Cells from the selected monoclonal stable SH-SY5Y cell line expressing HiBit-tagged tau were incubated in complete medium in the presence of 0.1% DMSO, 100 nm bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for 4, 8 or 12 h. The split luciferase complementation assay was performed from 100 μl of cell culture medium using the Nano-Glo HiBit Extracellular Detection System kit, according to the manufacturer’s instructions (N2421, Promega). This protocol is based on the established concept of bimolecular fluorescence complementation and detects extracellular tau luciferase activity only after complementation in the presence of extracellular LgBiT. Values were normalized to control samples treated with DMSO at 4 h. This experiment was performed in triplicate.
Cell death analysisLDH release was monitored from cell culture medium fractions collected at the indicated times to evaluate the levels of plasma membrane damage and cell death using a commercial LDH assay kit (ab65393, Abcam), according to manufacturer’s recommendations. Each experiment was performed in triplicate.
ELISA analysis of cathepsin D in cell mediumLevels of cathepsin D released into the cell medium were measured by ELISA. SH-SY5Y cells were incubated in complete medium in the presence of 0.1% DMSO, 100 nM bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for the indicated durations. Cathepsin D secretion levels in the cell medium were monitored using a commercial Cathepsin D ELISA kit (ab119586, Abcam) according to manufacturer’s recommendations. Values were normalized to control samples treated with DMSO at 4 h. This experiment was performed in triplicate.
Analysis of vesicular pHSH-SY5Y cells were incubated in complete medium in the presence of 0.1% DMSO, 100 nM bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for 6 h, and change in lysosomal pH was analyzed using LysoSensor Yellow/Blue DND-160 (L7545, Thermo Fisher Scientific). After drug treatment, cells were incubated with 2 µM LysoSensor DND-160 for 5 min in the presence of drugs and washed with probe-free medium, and overall cell fluorescence was measured using a SPARK multimode microplate reader with SPARKCONTROL Method Editor software (V 3.0, TECAN Trading). The ratio of blue and yellow intensities was quantified and is represented as fold change compared with control samples treated with DMSO. This experiment was performed in triplicate.
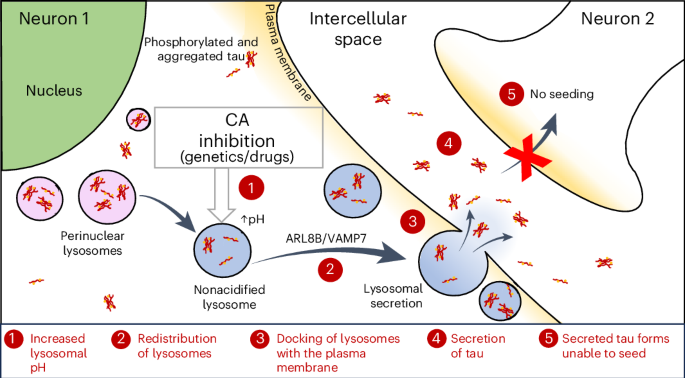
Analysis of lysosomal distributionSH-SY5Y cells (ECACC, 94030304) were cultured on coverslips and incubated in complete medium in the presence of 0.1% DMSO, 100 nM bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for 6 h. After treatment, cells were washed with PBS three times and fixed and permeabilized with ice-cold methanol for 4 min. After washing three times with PBS, cells were blocked with 3% bovine serum albumin (BP1605-100, Thermo Fisher Scientific) for 1 h. Cells were incubated with rabbit anti-LAMP1 (1:500; 9091, Cell Signaling Technology) in blocking buffer overnight at 4 °C. After washing three times with PBS, cells were incubated with secondary goat anti-rabbit Alexa Fluor 594 (1:500; A11012, Thermo Fisher Scientific) for 1 h at room temperature. Cells on coverslips were washed with PBS and mounted in ProLong Diamond Antifade Reagent with DAPI (P36962, Thermo Fisher Scientific). Imaging was conducted with an LSM880 Carl Zeiss confocal microscope with a ×63 oil-immersion lens using Leica Application Suite X microscope imaging software. Quantification of the distribution of LAMP1+ puncta was performed using CellProfiler Analyst (Broad Institute61). Briefly, for all images analyzed, the area of each nucleus and each cell was extracted using the module ‘MeasureObjectSizeShape’. An area of 5 μm width was then defined around the nucleus, allowing for the establishment of perinuclear and peripheral zones in each cell. The LAMP1+ signal in each area was assessed to determine whether lysosomes were mainly perinuclear or peripheral. A minimum of 40 cells per condition for each independent experiment were analyzed.
Cell transfection with siRNACells of the monoclonal stable SH-SY5Y cell line expressing HiBit-tagged tau and inducible SH-Tau cells (eGFP–tau-P301L) were transfected with specific individual siRNAs targeting VAMP7 and ARL8B using Lipofectamine 2000 transfection reagent (12566014, Thermo Fisher Scientific) following the manufacturer’s recommendations. Individual siRNAs were purchased from Dharmacon. The following siRNA sequences were used: scramble 5′-UGGUUUACAUGUCGACUAA-3′, ARL8B 5′-GAUGAGAAACAGCUAAUUG-3′ and VAMP7 5′-GUACUCACAUGGCAAUUAU-3′. After 3 days, cells were incubated in complete medium in the presence of 0.1% DMSO, 100 nM bafilomycin A1, 30 μM methazolamide or 30 μM methocarbamol for an additional 12 h.
Knockdown efficiency of targeted genes was monitored by western blotting. Briefly, 3 days after siRNA transfection, cell lysates were resolved by SDS–PAGE and transferred onto PVDF membranes. PVDF membranes were blocked with 4% skim milk in PBST for 1 h and incubated overnight at 4 °C with the following primary antibodies diluted in 4% skim milk PBST: rabbit polyclonal anti-ARL8B (1:1,000; ab207697, Abcam), rabbit polyclonal anti-VAMP7 (1:1,000; SAB2105695, Sigma-Aldrich) and rabbit anti-β-actin (1:1,000; A2066, Sigma-Aldrich). After several washes in PBST, membranes were incubated for 90 min at room temperature with HRP-linked anti-rabbit (7074, Cell Signaling Technology; 1:5,000) in 4% skim milk and 0.1% Tween-PBS. ARL8B, VAMP7 and β-actin bands were detected using the ECL enhanced chemiluminescence detection kit from GE Healthcare (RPN2106).
Tau seeding assayInducible SH-Tau cells (eGFP–tau-P301L) were cultured in X2 140-mm dishes as described in ‘Analysis of tau clearance in cell culture’. Cells were reverse treated (that is, tau expression was switched on and drug treatment was applied at the same time) by the addition of doxycycline (0.2 μg ml–1) in combination with methocarbamol or methazolamide at concentrations of 30 μM and DMSO as a control. Low FBS (1%) and low penicillin–streptomycin (10 U ml–1) DMEM/F12 medium was used for reverse treatment. The cells were incubated at 37 °C and 5% CO2 for 12 h. After 12 h, the medium was collected and concentrated using Amicon Ultra-15 Centrifugal Filters 10K (UFC901024, Millipore) to a volume of 300 µl; OptiMEM (Thermo Fisher Scientific) was added to increase the volume to 550 µl. Tau levels were determined using a Tau ELISA kit (KHB0042, Invitrogen) according to manufacturer’s instructions.
Seeding assays were performed largely as described previously29. HEK293 cells expressing tau-P301S–Venus were plated at 15,000 cells per well in poly-d-lysine-coated black 96-well plates (Corning, 3603) in 50 μl of OptiMEM (Thermo Fisher Scientific). Samples of medium containing released tau–GFP or recombinant tau-P301S assemblies at comparable concentrations were incubated with Lipofectamine 2000 (Invitrogen, 11668019) at a final concentration of 0.5 μl 50 μl–1 for 10 min at room temperature. Fifty microliters of the tau-containing medium sample per well was added for 1 h at 37 °C before the addition of 100 μl of complete DMEM per well to stop the transfection process. Cells were incubated at 37 °C for 72 h after DMEM addition. Tau–Venus aggregates were imaged and quantified using a Nikon Ti2 ECLIPSE and high-content imaging analysis software (NIS-Elements AR V. 5.41.02 Nikon) and calculated using the following equation:
$$}\,}=(}\; }\; }\; }\;/\;}\; }\; })\times 100.$$
Mouse experimentsMouse studies were performed in accordance with the UK Animals (Scientific Procedures) Act with appropriate Home Office Project and Personal animal licenses and with University of Cambridge Animal Welfare and Ethical Review Body approval. Mice were maintained and used in experiments following PREPARE and ARRIVE guidelines.
MiceMice were housed in individually ventilated cages with free access to standard animal food chow and water in a climate-controlled room (45–65% humidity and 20–24 °C) on a 12-h light/12-h dark cycle. We used two different neurodegenerative disease mouse models. The frontotemporal dementia (Tg4510) mouse model, overexpressing the human tau 0N4R isoform carrying the P301L mutation, as described previously1,62, was generated by crossing two transgenic lines. Both congenic parental mouse strains, tetracycline-controlled transactivator (tTA) Tg(Camk2a-tTA)1Mmay (007004) and hTau responder FVB-Tg(tetO-MAPT*P301L)#Kha/JlwsJ (015815), were purchased from The Jackson Laboratory. Tg4510 mice were bred from these two different parental lines to activate the expression of transgene under the control of a tetracycline conditional gene expression system (tet off, tTA). Transgene expression is largely restricted to the forebrain by the Camk2a promoter. Although the mice can be fed with a doxycycline-supplemented diet (R105 with 200 ppm doxycycline, SAFE Diet) to switch off transgene expression, in the current study, we did not repress transgene expression (that is, it remained switched on). The second disease model was the tau (PS19) mouse model, B6N.Cg-Tg(Prnp-MAPT*P301S)PS19Vle/J (024841, The Jackson Laboratory). This mouse model expresses the human tau 1N4R isoform with the P301S mutation driven by the mouse prion protein promoter32.
Methazolamide administration Injection (i.p.) and OGA stock solution of methazolamide (sc235615, Santa Cruz Biotechnology) was prepared at 200 mg ml–1 in DMSO. This was diluted to a final formulation to administer to 3- to 4-month-old wild-type C57BL/6J mice according to body weight with 2% Tween 80 and 30% PEG300 in saline (freshly prepared) on the day. As a control, a separate group of mice was administered vehicle only with 5% DMSO instead of drug with 2% Tween 80 and 30% PEG300 in saline.
Subcutaneous administration by osmotic minipumpA solution of methazolamide in DMSO with 30% PEG300 and 2% Tween 80 was given subcutaneously as a continuous infusion with a daily dose of 10, 20 or 50 mg per kg (body weight) by implanting osmotic minipumps (0.5 μl h–1 flow rate; Alzet Model 2002) in 3.5- to 4-month-old Tg4510 and 8- to 9-month-old PS19 mice. The concentration of methazolamide in the minipump was adjusted according to each animal’s body weight to obtain the required delivery of 10, 20 or 50 mg per kg (body weight) per day, and the pumps were primed by soaking in saline at 37 °C overnight. Minipumps were replaced after 14 days with freshly loaded minipumps. As a control, a separate group of mice was administered vehicle only (that is, DMSO, 30% PEG300 and 2% Tween 80) via minipump.
Pharmacokinetic analysisWild-type (C57BL/6J) male mice (3–4 months old) were administered methazolamide (200 mg per kg (body weight)) either by i.p. injection or OG. Mice were anesthetized using isoflurane at eight different time points (that is, 5, 10, 15, 30, 60, 120, 240 and 480 min), with three mice dosed for each time point. Once a mouse was anesthetized, a terminal blood sample was taken by cardiac puncture. Blood (0.3–0.8 ml) was collected in EDTA tubes (Microvette 100K3E, Sarstedt). The plasma fraction was immediately separated by centrifugation (1,000g, 5 min, 4 °C) and stored on dry ice and then −80 °C until liquid chromatography–mass spectrometry analysis to determine drug concentration. After confirming death by cervical dislocation, the brains were collected, frozen on dry ice and stored at −80 °C until processing.
Repeat blood sampling (via the saphenous vein) was also performed during the course of continuous dosing of methazolamide via osmotic minipump according to the experiment (at day 13 for Alzet Model 2002), followed by collection of a terminal sample at day 28. At each occasion, blood samples of 100–120 μl were collected in EDTA tubes (Microvette 300K2E, Sarstedt). The plasma fraction was separated and stored as described above.
Drug concentration and pharmacokinetic analyses were performed on plasma and brain tissues using liquid chromatography–mass spectrometry and Noncompartmental Pharmacokinetic Data Analysis software (Windows 2.0.6 Excel 2002 Edition) for pharmacokinetic analyses by Q3 Analytical.
CA activityCA activity was measured by a colorimetric-based assay using a BioVision Carbonic Anhydrase Activity Assay kit (K472-100) according to manufacturer’s instructions with modifications. Twenty to 40 mg of tissue was excised from the cortical region of mouse frozen brains with a scalpel, transferred into a tube and thawed on ice in 1 ml of PBS to clean the tissue. PBS was removed and replaced with 10 μl per mg of tissue of ice-cold 1 mM Tris (pH 8.0) and 200 mM NaCl, and eight glass beads were added to facilitate tissue disruption. After homogenization by sonication (three cycles, 20 s each), samples were centrifuged at 16,900g for 15 min at 4 °C. The supernatant was collected and diluted 1:10 in 1 mM Tris (pH 8.0) and 200 mM NaCl for measuring protein concentration by the fluorometric Qubit assay (Qubit, Thermo Fisher Scientific). For plasma samples, plasma was thawed on ice and centrifuged at 3,000g for 1 min at 4 °C to remove any cells. Clear plasma was then diluted 1:10 in 1 mM Tris (pH 8.0) and 200 mM NaCl for protein concentration assays. Due to the technical difficulties in collecting enough blood sample for this assay, the enzymatic assay from plasma could not be performed on all three mice for each time point used in the pharmacokinetic study.
To measure CA activity in brain homogenates and plasma, 10 μg of protein in 10 μl was added to each well, with samples analyzed in duplicate. Eighty-five microliters of CA buffer was added to each well (provided in the kit), and the plate was incubated at room temperature for 15 min. A standard curve with increasing concentrations of nitrophenol from 0 to 40 nmol per well (provided in the kit) was run in parallel in duplicate. After incubation, 5 μl of substrate (provided in the kit) was added to each sample well, and absorbance (405 nm) was measured immediately after the addition and every 5 min subsequently for 1 h at room temperature in a Elx800 plate reader (Appleton Woods). CA values were normalized to the activity in untreated/placebo mice.
Tau sarkosyl extraction from mouse brainBrains from PS19 mice underwent soluble and insoluble fractionation using a sarkosyl extraction protocol1. Cerebral cortex from mouse brain tissue was homogenized by using a Precellys CK14 Lysing kit (10144-554, Avantor) in TBS buffer (20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EGTA and protease inhibitor cocktail). Protein in mouse brain lysates in TBS buffer was quantified by bicinchoninic acid assay. After the addition of Laemmli buffer, samples were boiled for 5–7 min at 100 °C. Western blots for both mouse sarkosyl-soluble and sarkosyl-insoluble fractions and brain lysates were performed on 10% acrylamide SDS–PAGE gels. The same vehicle-treated control mouse sample (for the insoluble fraction) was loaded on each gel for normalization and to allow the comparison between the bands detected on the two different membranes. Blots were blocked in 5% nonfat milk in PBST and incubated with the following primary antibodies overnight: mouse anti-Tau5 (1:1,000; ab80579, Abcam), mouse anti-PHF1 (1:1,000; a kind gift from P. Davies, Albert Einstein College of Medicine, New York, USA) and rabbit anti-GAPDH (1:5,000; NB100-56875, Novus Biologicals). The membranes were labeled with fluorescent secondary antibodies and were analyzed with a LI-COR-Odyssey CLX apparatus. Densitometry analysis on the immunoblots was performed using IMAGE STUDIO Lite software.
Novel object recognition testA novel object recognition test, commonly used to assess changes in short-term memory tasks, was performed as previously described63. Behavioral video recording in mice was performed using a Logitech C310 HD Webcam camera connected to Panlab Smart Video Record It Software (Smart 3.0) and analyzed manually using Behavioral Observation Research Interactive Software (BORIS version 8.25.4)64.
Statistical analysis on the percentage of time spent on a novel object was performed using a Wilcoxon matched-pairs signed-rank test. All analyses were performed using Prism (GraphPad).
Immunohistochemistry of mouse brainsMouse brain immunostaining and immunohistochemical analysis was performed as described in ref. 63.
Stereologic analysis to quantitate neuron loss in the CA1 region in a statistically ‘unbiased’ fashion was performed using previously described techniques1,
留言 (0)