The work has been reported in line with the ARRIVE guidelines 2.0.
Cell lines and culture
The cervical squamous carcinoma cell line SiHa and cervical adenocarcinoma cell line HeLa were acquired from Shanghai Cell Biology Medical Research Institute, Chinese Academy of Sciences. The normal cervical epithelial cell line ECT1 was obtained from Shanghai BinSui Biological Technology Co., Ltd. High-glucose Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin-streptomycin solution (Pen-Strep, 100 mg/mL; Invitrogen, UK) was employed to cultivate SiHa and HeLa cells. And Eagle’s minimum essential medium (EMEM) was used to incubate ECT1 cells. Cells were cultured at 37 °C in a humidity-controlled atmosphere with 5% CO2.
Compounds
Cisplatin (DDP, #HY-17394; MCE, New Jersey, USA) was solubilized in distilled water to generate 2 mM solution and preserved at -20 °C, which was diluted to specific concentrations with complete DMEM medium before use.
Separation and incubation of hCD-MSCs
The separation and incubation of hCD-MSCs were performed based on our previously mentioned protocol [15]. Placental chorionic tissues from healthy and full-term pregnant women (25–30 years of age) were obtained following cesarean section, which received approval from ethics committee of the Second Affiliated Hospital of Wenzhou Medical University. The chorionic tissues were washed with phosphate-buffered saline solution (PBS) and chopped up completely, and subsequently dissociated with 1 mg/mL collagenase type II (Solarbio, China) for 1 h at 37 °C in a water-bath shaker. Thereafter, cell suspension underwent filtration utilizing a 100-µm nylon mesh cell filter to remove the tissue and cellular debris. Next, cell suspension was gathered and subjected to centrifugation at 1000 rpm for 10 min, and the supernatant was removed carefully. Afterwards, isolated hCD-MSCs were resuspended in low-glucose DMEM medium consisting of 10% FBS and 1% Pen-Strep. The cell culture medium was replaced at 2-day intervals and cells were passaged at a confluence density ranging from 80 to 90%.
Multipotent differentiation and immunophenotype characterizations of hCD-MSCs
The hCD-MSCs at passage 3 (P3) were selected to for multipotent differentiation and immunophenotype characterizations. An appraisement of the multipotency of hCD-MSCs towards adipogenic, osteogenic, and chondrogenic lineages was performed. Concisely, cells were inoculated in 24-well plates at a density of 5000 cells per well. When cell confluence reached 60-70%, cell culture medium was replaced with adipogenic differentiation medium (Gibco, USA), osteogenic differentiation medium (Gibco, USA), and chondrogenesis differentiation medium (Gibco, USA), respectively. 4% paraformaldehyde fixation for 30 min was administrated in cells 2 weeks later. Subsequently, cells were subjected to Oil red O staining (Solarbio, China), Alkaline phosphatase staining (Beyotime, China), and Alcian blue staining (Solarbio, China), respectively, for confirming the positive induction.
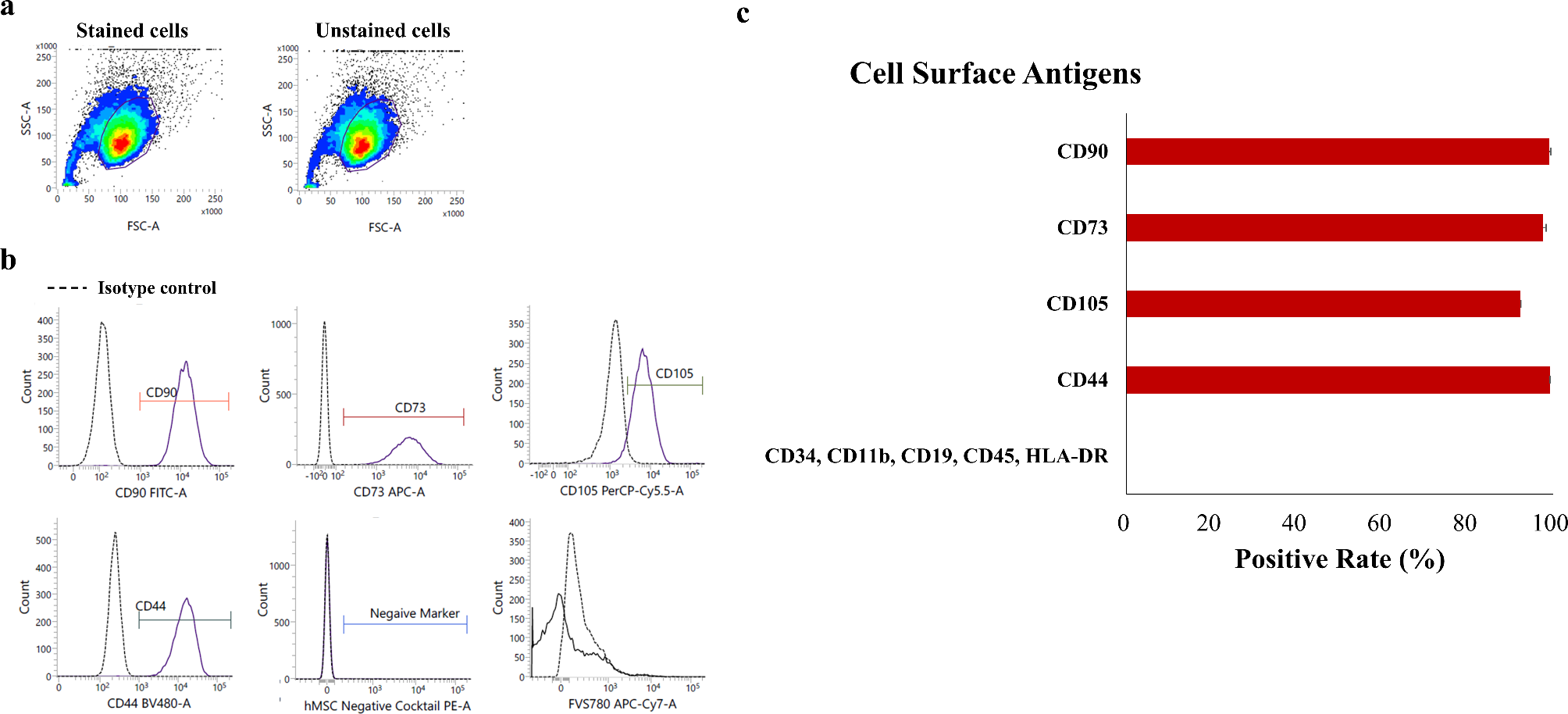
The immunophenotype of hCD-MSCs was characterized by flow cytometry with fluorescent-labeled monoclonal antibodies against CD90, CD73, CD44, CD45, CD34, HLA-ABC, and HLA-DR (ThermoFisher, USA). Cells were subjected to trypsinization and PBS washing, followed by incubation with respective antibodies for 20 min at room temperature in light-protecting condition. Afterwards, the samples were processed by CytoFLEX flow cytometry (Beckman Coulter, Fullerton, USA).
Immunohistochemistry (IHC)
The cervical cancer tissues and paired adjacent non-cancerous cervical tissues were incubated with primary antibody DR4 (1:200, Affinity Biosciences, #AF0304) following the guidelines of immunohistochemistry. Concisely, deparaffinized and rehydrated tissue sections were subjected to microwave oven for antigen retrieval. Next, the tissue sections were incubated with primary antibodies overnight at 4 ℃ followed by incubation with appropriate secondary antibodies. A freshly-prepared DAB solution was employed for chromogenic reaction and nuclei were counterstained with hematoxylin. Positive staining appeared in brown, and representative pictures were captured by microscope (Leica Microsystems, Wetzlar, Germany).
Western blotting assay
Total proteins were extracted from cell lysates and phenylmethanesulfonylfluoride (PMSF) was applied to prevent proteins from degradation. The protein quantification was done by bicinchoninic acid (BCA) protein assay kit (Beyotime, China). The prepared protein (40 µg/path) was resolved on polyacrylamide sodium dodecyl sulfate (SDS-PAGE) gels and subsequently electro-transferred onto polyvinylidene difluoride (PVDF) membranes. Following blocking with 5% milk, membranes were incubated with primary antibodies overnight at 4 ℃. Primary antibody of DR4 (1:2000, Affinity Biosciences, #AF0304) were employed and the anti-rabbit secondary antibodies conjugated with horseradish peroxidase (HRP) were employed to detect the protein bands. The protein bands were visualized with enzyme-linked chemiluminescence detection kit (ECL) under the Chemiluminescence Imaging System (ChemiScope 6000, CLiNX, China).
Lentivirus and transfection
The pL-CMV-GFP-TRAIL-LAMP2b lentiviral vector and the negative control vector pL-CMV-GFP-blank-LAMP2b were acquired from Shanghai Nuobai Biological Technology Co., Ltd. (China), which were co-cultured with HEK-293T cells by using Lipofectamine 2000 (Invitrogen, USA) transfection reagent with a lentivirus transfection system, respectively. Then, the resulting virus particles were applied to transfect hCD-MSCs. The cell model stably overexpressing TRAIL (hCD-MSCsTRAIL) was generated by selection with 10 µg/mL Blasticidin-S (Solarbio, China), and the protein expression of TRAIL (1:1000, CST, #3219) was examined by western blotting.
Exo extraction and characterization
To obtain hCD-MSCs and hCD-MSCsTRAIL secreted Exo, cells were first maintained in low-glucose DMEM medium consisting of 10% FBS and 1% Pen-Strep until cell density reached 60–70% confluence. Cells were subsequently cultured in low-glucose DMEM medium containing 10% exosome-free FBS and 1% Pen-Strep for an additional 48 h. Then, culture supernatant was collected followed by a differential centrifugation at 4℃, including 3,000 g for 10 min and 10,000 g for 30 min to eliminate cells and debris. Exo was extracted at 100,000 g for 90 min at 4℃ in a Type Ti32 rotor by ultracentrifugation (Beckman Coulter, USA). After meticulously eliminating the supernatant, Exo was washed in PBS followed by a 90 min spin at 100,000 g in a Type Ti41 rotor at 4℃, and ultimately resuspended in PBS for preservation at -80℃ for follow-up usage. Herein, Exo derived from hCD-MSCs and hCD-MSCsTRAIL were referred as hCD-MSCs-Exo and hCD-MSCs-ExoTRAIL, respectively.
The morphology of Exo was visualized and photographed with transmission electron microscopy (TEM; Tecnai G2 Spirit, Fei, USA). The size distribution of Exo was discerned by nanoparticle tracking analysis (NTA; ZetaView PMX 110, Particle Metrix, Germany). The protein content in Exo was determined by BCA protein assay according to manufacturer’s instructions. Moreover, representative markers (Alix, Annexin-V and Flotillin-1) and negative marker (GM130) of Exo were identified by Western blotting. Primary antibodies against Alix (1:1000, CST, #2171), Annexin-V (1:1000, CST, #8555), Flotillin-1 (1:1000, CST, #18634) and GM130 (1:1000, CST, #12480) were employed. The corresponding secondary antibodies conjugated with horseradish peroxidase (HRP) were visualized to inspect the protein bands.
Uptake of Exo in cervical cancer cells
The purified hCD-MSCs-Exo and hCD-MSCs-ExoTRAIL were labeled with PKH26 red fluorescent labeling kit (Umibio, China). Briefly, Exo suspension was mixed with 100 µM PKH26 staining working solution at room temperature for 30 min shielded from light, and the reaction was ceased by adding 100 µL low-glucose DMEM medium containing 10% exosome-free FBS. The PKH26-labeled Exo was resuspended with PBS and centrifuged at 100,000 g in a Type Ti41 rotor for 90 min at 4 ℃ to remove the excess dye. The collected Exo were named as PKH26-hCD-MSCs-Exo or PKH26-hCD-MSCs-ExoTRAIL.
The cervical cancer cells (SiHa and HeLa) were seeded onto coverslips in 6-well plates (2 × 105 cells/well) and cultured for 24 h at 37°C with 5% CO2. Afterwards, cells were grown with medium consisting of 4 µg/mL PKH26-hCD-MSCs-Exo or 4 µg/mL PKH26-hCD-MSCs-ExoTRAIL for 24 h shielded from light, at 37°C with 5% CO2. Subsequently, cells were treated with PBS rinsing, 4% paraformaldehyde fixing, and 4’, 6-diamidino-2-phenylindole (DAPI; Abcam, ab104139) dyeing. Eventually, cells were photographed with the laser scanning confocal microscope (Leica, Germany).
Cell viability assay
Cells were inoculated at a proper density in 96-well plates overnight and subsequently incubated with different vehicle or therapeutic regimens for 48 h. Thereafter, CCK-8 reagent (10 µL/well) was added for another incubation of 2 h, and the optical density (OD) at 450 nm was acquired with Micro-plate Reader (Bio-Rad, USA).
Cell apoptosis assay
Cells were inoculated at a proper density in 60-mm dish overnight and subsequently incubated with vehicle or therapeutic regimens for 48 h. Next, cells were gathered through trypsinization, resuspended in binding buffer, and double stained with Annexin V conjugated with phycoerythrin (PE) and 7-amino-actinomycin D (7-AAD) for 20 min avoiding light. Stained cells were evaluated with CytoFLEX flow cytometry (Beckman Coulter, Fullerton, USA).
Fabrication of DDP & hCD-MSCs-ExoTRAIL
To entrap drug DDP into hCD-MSCs-ExoTRAIL, 32 µg/mL of hCD-MSCs-ExoTRAIL and 6 µM of DDP were mingled with ice-cold PBS in 0.4 cm electroporation cuvette (Bio-Rad, USA). Afterwards, the mixture was electroporated by once pulse at 400 V, 150 µF capacitance using Gene Pulser Xcell (Bio-Rad, USA), and subsequently maintained at 37 ℃ for 30 min for the recovery of Exo membrane. Following the reaction termination, the mixture was ultracentrifugated at 100,000 g for 90 min to eliminate free DDP and obtain DDP & hCD-MSCs-ExoTRAIL nanoparticles. The platinum encapsulated in Exo was quantified using inductively coupled plasma mass spectrometry (ICP-MS) and DDP loading efficiency (%) in DDP & hCD-MSCs-ExoTRAIL nanoparticles was calculated. The traits of the fabricated DDP & hCD-MSCs-ExoTRAIL nanoparticles were described with TEM and NTA. For stability evaluation, nanoparticles were dispersed in pure PBS at -80 ℃ or 4 ℃. And the size distribution of nanoparticles was measured by dynamic light scattering (DLS) using a nanoparticle size analyzer at the days 0, 1, 2, 3, 5, 7.
Drug administration in tumor-bearing nude mice model
All animal experiments were in line with the regulations promulgated by the Institutional Animal Care and Use Committee of Wenzhou Medical University (No.wydw2021-0176). Female BALB/c nude mice (5 weeks of age) were ordered from Beijing Vital River Laboratory Animal Technology Co., Ltd., and housed in standard rearing condition. To establish cervical cancer-bearing mice, 1.5 × 106 SiHa cells in 100 µL PBS were transplanted subcutaneously in the right buttock of mice aged 7 weeks. When tumors grow to around 30 mm3 (volume = ½ × length × width2, as examined with vernier caliper [16]), mice were randomized into 4 clusters with 4 mice per cluster and subsequently intratumorally administrated with PBS, DDP (2 mg/kg), hCD-MSCs-ExoTRAIL (2 µg/g), and DDP & hCD-MSCs-ExoTRAIL (as obtained from electroporation with 2 mg/kg DDP and 2 µg/g hCD-MSCs-EXOTRAIL), respectively. The administration was repeated at 4-day intervals for 4 cycles. The tumor size and mice body weight were monitored before administration every time, and the last measurement was done 4 days after the fourth treatment.
Four days post the fourth treatment, mice were anesthetized intraperitoneally with pentobarbital sodium (100 mg/kg, #P3761; Sigma, USA). Peripheral blood collection of mice via submandibular vein puncture was applied for blood routine and biochemistry inspection. Whereafter, mice were euthanized by cervical dislocation method, and major organs (heart, liver, spleen, lung, kidney) and tumors were harvested, 4% paraformaldehyde-fixed, dehydrated, paraffin-embedded and sectioned. Tissue sections of major organs were deparaffinized and subjected to hematoxylin-eosin (HE) staining for histopathological lesions assessment. Tumor slices were processed for fluorescent terminal-deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) staining to evaluate tumor cells apoptosis. Additionally, total proteins were extracted from frozen tumor tissues, and protein expressions of JNK (1:1000, abcam, #ab179461), c-Jun (1:1000, CST, #9165), p-c-Jun (1:1000, CST, #3270) and survivin (1:1000, CST, #2808) in tumors were detected by Western blotting.
Statistical analysis
Experimental data were analyzed and plotted with GraphPad Prism 8.0 software, and delineated as mean ± standard deviation (SD). Statistical criteria were validated by Student’s t-test for two groups and One-way analysis of variance (ANOVA) for three or more groups. The least significance method was employed for data exhibiting homogeneous variances, and data with nonhomogeneous variances were analyzed with Dunnett’s T3 method. Moreover, non-parametric data were compared by Mann-Whitney U tests for two groups and Kruskal-Wallis tests for three or more groups. Significant probability (P)-values are represented as****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05.







留言 (0)