
記住我



To build a developmental-status-aware, tissue-specific cell-state reference for various human tissues, we compiled a series of single-cell RNA-seq data from both fetal and adult samples in the Human Cell Landscape (HCL) cohort [19]. We first identified significantly over-expressed genes in each of the annotated cell types and then used them to build a signature matrix, as described previously (Additional file 2: Table S2) [23, 24] (Fig. 1A). To reduce the bias that might confound the decomposition process, we excluded genes involved in the cell cycle, ribosome biogenesis, cell apoptosis, and mitochondrial genome. In this study, we focused on seven major human solid tissues where both fetal and adult single-cell data were available, namely the adrenal gland, brain, kidney, liver, lung, pancreas, and stomach. Through a UMAP [31] projection of the signature matrices, we found that fetal cell types were globally distinct from the adult cell types in each tissue, even in cases where a fetal cell type and its adult counterpart were essentially the same cell type (e.g., fetal and adult macrophages), indicating that the developmental status is a major determinant of cellular gene expression programs (Additional file 3: Fig. S1A–G). Importantly, the observed fetal-adult separation was not due to a sample-specific batch effect, as the HCL cell types from different biological replicates (donors) were clustered by the developmental stage rather than their sample origin (Fig. S1H). Finally, given a specific tissue, we decomposed bulk RNA-seq data based on the corresponding single-cell expression signatures using CIBERSORT [23, 24] to obtain the relative strength of individual cell state programs (Fig. 1A).
Fig. 1
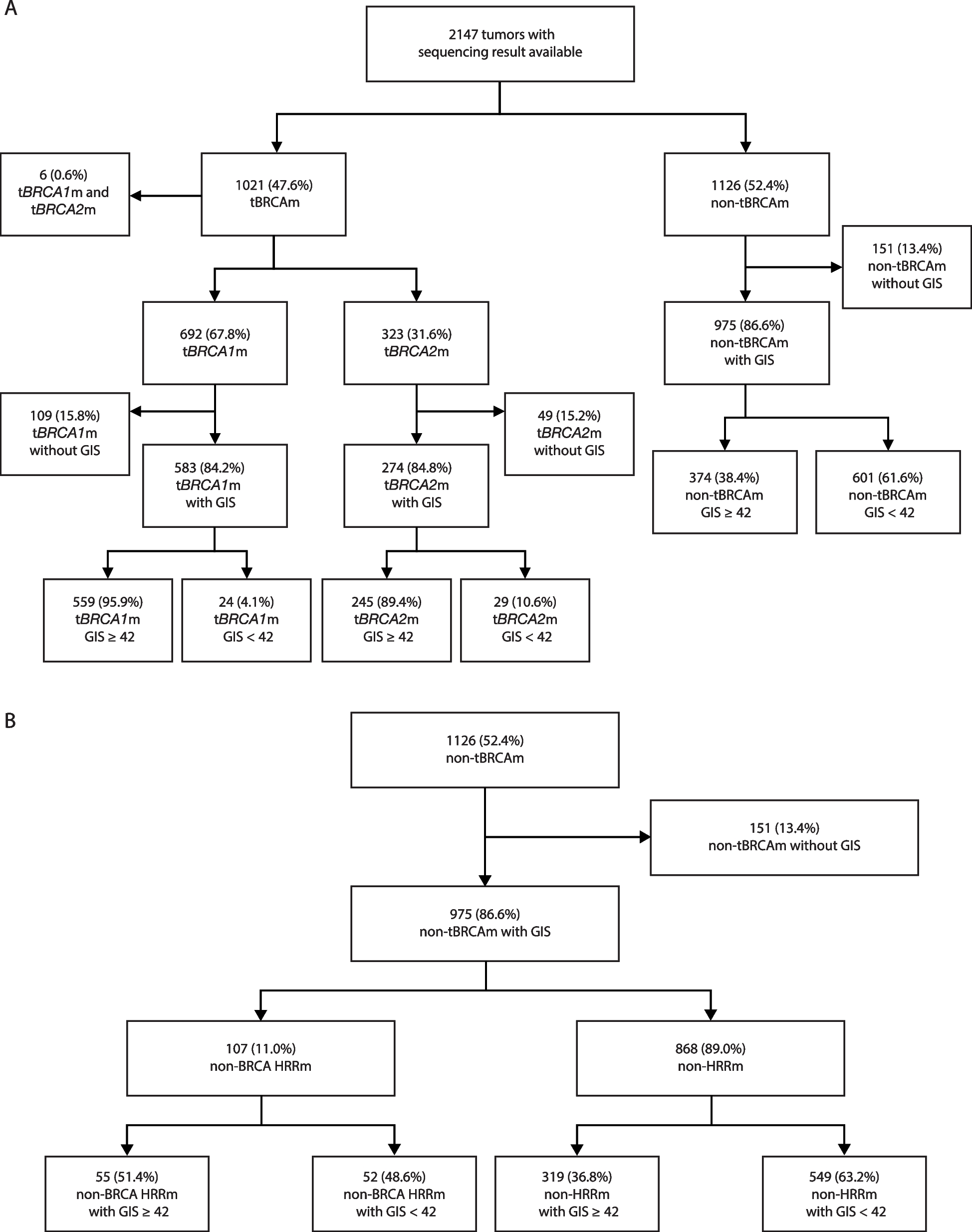
Overview and performance validation of a developmental-status-aware, tissue-specific decomposition strategy. A Schematic of the decomposition procedure. B–E Benchmarking of the decomposition approach using the EvoDevo tissue samples, including the human brain (B), cerebellum (C), kidney (D), and liver (E). Stacked bar plots in the top panels represent the relative fractions of cell types. An uncolored gap is added to separate the fetal and adult components. For each tissue, cell types with a maximum relative fraction of < 5% across all the samples are combined and shown as a single bar at the bottom, separated from others with an additional gap. Trendlines in the bottom panels show the changes in total fetalness along the developmental trajectory. Error bars denote 95% confidence intervals
The ontogenesis of a human organ into its adult stage is characterized by an extensive turnover of a myriad of cell types, along with the transformation of gene expression programs within each cell type. We hypothesized that our decomposition strategy could effectively encapsulate such complex and dynamic cellular compositions from a series of bulk gene expression profiles. To test this hypothesis, we collected RNA-seq data from the EvoDevo study [32], where the complete developmental trajectories of the human brain, cerebellum, kidney, and liver tissues were well characterized. Based on this gold standard, we assessed whether the developmental dynamics could be accurately recovered. Indeed, our decomposition results (i) captured the global trend of a gradual loss of fetal cell types, (ii) identified the transition point exactly matching the newborn in each tissue, and (iii) provided detailed molecular portraits of specific cell types that are well-aligned with established findings (Fig. 1B–E). For example, in the brain tissue, there were barely any neurons at five weeks post-conception (wpc), the bulk being composed of neuronal progenitors such as the radial glia (Fig. 1B), consistent with the fact that neuronal production does not start until six wpc [33]. At seven wpc, the tissue swiftly transforms into a fetal-neuron-rich compartment, and after birth, it eventually becomes a fully functional ecosystem with a much higher cellular diversity where astrocytes, oligodendrocytes, neurons, glia cells, and progenitors co-exist (Fig. 1B). Similarly, in the liver, a transition from erythroid progenitors into erythroid cells occurs at seven wpc and erythroid cells are then maintained as the dominant cell type in the fetal liver until at least 20 wpc (Fig. 1E), corroborated by a recent time-course scRNA-seq study on the human fetal liver [34]. These results confirmed the accuracy of our decomposition approach in capturing the developmental stage of cellular composition transformation from bulk RNA-seq data.
Revealing cellular heterogeneity of human adult tissues at a high resolutionTo demonstrate the capacity of our decomposition strategy to reveal the detailed cellular composition of human tissues, we decomposed > 4000 human samples of seven adult tissues from a cohort of a much larger scale, the Genotype-Tissue Expression (GTEx). We successfully characterized the cellular composition of these tissues, largely matching classical anatomic and physiological observations (Fig. 2A–G). For example, we found that the adrenal gland samples were mainly composed of zona fasciculata cells (Fig. 2A), which are known to occupy the middle and also the largest zone of the adrenal cortex [35]. The second most common cell type was the intra-adrenal chromoblast, which serves as the progenitor to the chromaffin cells [36] (not annotated by the HCL study) that take up most of the adrenal medulla [37]. Likewise, we inferred that the most common cell type (~ 80%) in the liver was hepatocytes (Fig. 2C), highly consistent with the anatomical estimation (80%) of this cell type in the tissue mass [38]. The second common cell type was the sinusoidal endothelial cell, which represents ~ 15% of the liver cells [39].
Fig. 2
Cell-state decomposition of human adult normal tissues. A–F Stacked bar plots showing the fractions of cell types inferred in 6 GTEx tissues, i.e., adrenal gland (A), kidney (B), liver (C), lung (D), pancreas (E), and stomach (F). An uncolored gap is added to separate the fetal (top) and adult components (bottom). Samples are ordered by the total proportion of inferred tissue-specific fetal cell types, from the smallest to the largest. The top two fetal and adult cell types with the highest relative fractions are highlighted. G Violin plots showing the relative fractions of 8 brain cell types across the 13 GTEx brain sub-regions and two EvoDevo brain cohorts. H Bar plots showing Spearman’s rank correlation coefficients between total adult cell fractions and individual age across 13 GTEx brain sub-region cohorts. Statistically significant correlations (FDR < 0.05) are indicated with asterisks. I Correlation of estimated fractions of all 22 brain cell types between the GTEx cerebellar cohort (including the cerebellum and cerebellar hemisphere) and the EvoDevo cerebellum cohort. The translucent band around the regression line indicates the 95% confidence interval for the regression estimated through a bootstrap. J Same as I but for GTEx cerebral cohorts (including the cortex and frontal cortex [BA9]). K UMAP projection of GTEx brain sub-region samples based on inferred relative cell fractions. L UMAP projection in K overlaid with another UMAP projection of the EvoDevo brain samples. Grey circles indicate GTEx samples from K, which form the background to highlight EvoDevo samples
Notably, two observations seem to contradict the canonical views of adult tissue anatomy but can be well justified by recent literature. First, we detected fetal cell programs in all seven tissues, and in some cases, they composed a large proportion of the overall transcriptional state. For instance, fetal zona fasciculata cells constituted 12.4% (median) of the adrenal glands (Fig. 2A), which likely reflects the existence of a group of progenitor cells adopting a zona fasciculata phenotype, as reported in a study on a mouse model [40]. We found comparable cases, such as the presence of fetal distal tubule progenitors in the kidney (Fig. 2B), fetal pericytes in the lung (Fig. 2D), and fetal radial glia in the brain (Fig. 2G), all of which have been confirmed as sources for adult tissue maintenance and regeneration [41,42,43]. Thus, the presence of fetal cell types in the decomposition profiles suggests the intrinsic heterogeneity of human adult tissues and the inhabitation of stem or stem-like cells that naturally bear a resemblance to their fetal counterparts in their gene expression signature. Second, in contrast to the common view that the lung consists of mostly capillary endothelial cells and type I and type II pneumocytes [44], we estimated a considerable proportion of the lung mass to be resident macrophages (4.6–58.7%, median = 34.0%). Interestingly, a recent study using immune cell signatures to decompose samples across 46 GTEx tissues identified the lung as the organ with the highest macrophage infiltration [45]. Furthermore, a deeper, more comprehensive single-cell atlas study on the human lung revealed that macrophages were as abundant as 19.6–32.5% of the total lung mass [46]. This estimation is far above the classically determined abundance but very close to our decomposition.
To further assess the power of our decomposition strategy, we applied it to the GTEx brain cohort, where RNA-seq samples from 13 sub-regions were collected to allow a high-resolution, biologically informative investigation of cellular composition. We successfully recovered a series of signature cell types within each region, all supported by the literature. For instance, we noticed that although the brain sub-regions had variable cell compositions, the cerebral and cerebellar regions showed the sharpest contrast: the cerebellum was mostly enriched in excitatory neurons while largely depleted of oligodendrocytes that were prevalent in the cerebrum. Indeed, by directly counting the absolute numbers of various brain cells in mice, a recent study estimated that oligodendrocytes composed < 10% of the cerebellar mass and varied from 20 to 40% in other brain regions, but the neurons formed as much as 80% of the mouse cerebellum [47]. In another example, we detected a high abundance of interneurons in the hypothalamus, which is reportedly developmentally derived from interneurons in the alar plate [48]. Furthermore, we observed that Bergmann glia cells comprised a higher fraction of the cerebellum than other regions, well-aligned with the notion that they are “specialized astrocytes” in the cerebellum [49]. Finally, previous studies show that in the mouse brain, a few progenitor cell types declined during aging, although the global cellular composition remained largely unchanged [50,51,52,53]. Consistently, we detected moderate but significant positive associations between the total adult cell fraction and age in 10 of the 13 brain regions (FDR < 0.05, Fig. 2H).
To validate our decomposition results of the GTEx brain cohort more quantitatively, we decomposed a cohort of human brain RNA-seq profiles from the EvoDevo study [32]. We found that the estimations in the two independent cohorts were in full agreement both globally and locally, and the inferred cell compositions were very highly correlated (Fig. 2G, I, Rs = 0.86; Fig. 2L, Rs = 0.70). Intriguingly, while the adult brain samples were clustered together based on the cerebrum-cerebellum separation, the fetal samples almost exclusively formed a single tight cluster that was located closer to the adult cerebrum in the UMAP-projected space (Fig. 2L). This observation suggests that the prenatal brain, despite having committed to a cerebral or a cerebellar fate, maintains a shared gene expression program that undergoes dramatic divergence through postnatal development. Collectively, our decomposition approach demonstrated great power in revealing cellular heterogeneity from bulk RNA-seq data and further provided a high-resolution cellular composition map of human adult tissues in which transcriptionally fetal-like cell types appeared to be pervasive.
Resolving ambiguous cellular identities during cell state transitionsIn the above sections, our decomposition strategy showed an outstanding performance in quantifying the cell state composition of a bulk sample where the “ground truth” is known to some extent. We next aimed to assess the utility of this approach when a more flexible and dynamic definition of “cell state” is adopted, meaning that decomposition cell estimates may not have to reflect the heterogeneity of cell composition but represent the position of a mixture transcriptome (e.g., a tumor bulk) projected onto a cell state reference hyperspace where the developmental status is explicit. For this purpose, we applied our approach to in vitro cell culture systems under differentiation or de-differentiation, which bear two desirable properties. First, an in vitro cell culture is a mostly homogenous cellular community, and its cell state space is supposed to be constrained by the ground truth of the existent cell types. Second, such a system indeed experiences dramatic cell state transitions that are highly relevant to the tumor development process (Fig. 3A).
Fig. 3
Decomposition-based cell state anchoring in in vitro transitions. A Schematic of the rationale for using decomposition to resolve the ambiguity of cell identities in an in vitro culture system based on their gene expression profiles. B–M Trendlines show alterations of relative cell fractions during cell state transformation in induced differentiation or de-differentiation assays. D CPM, carboxypeptidase M, a marker of ventralized anterior foregut endoderm (VAFE) cells; AO, alveolar organoids; P0/2/5, passages 0, 2, and 5; SFTPC, surfactant protein C, a marker of alveolar stem cells. G iPSC, induced pluripotent stem cell; NPC, neural progenitor cell; MN, motor neurons. H PHC, primary hepatocyte; HepLPC, liver progenitor-like cell; TEM, transition and expansion medium. J GEC, gastric epithelial cell; hiEndoPC, human-induced endodermal progenitor cell. K HEP, hepatocyte; iHPC, induced hepatic progenitor cell; iHPCdf, re-differentiated induced hepatic progenitor cell. Error bars denote 95% confidence intervals
We compiled a series of in vitro time-course differentiation and de-differentiation RNA-seq profiles and applied our decomposition approach to them. We successfully recovered the major cell state programs in each culture system, along with the detailed alterations of their relative strength (skeletal muscle, neuron, AT2 cell, podocyte, ependymal cell, neuron, hepatocyte, β cell, gastric epithelial cell, hepatocyte, β cell, and distal progenitor cell in Fig. 3B–M, respectively). Moreover, our approach characterized the uncertainty of transient cell states by assigning the transcriptionally closest cell programs to the unknown/uncertain composition. For instance, in a case where lung AT2 cells were derived from iPSCs through lung progenitors [54], we observed the emergence of AT2 cell signals in the final stage of in vitro expansion, as originally reported. We were also able to clarify the identity of the intermediate cells as a mixture of the two major lung progenitor cell states, namely, the lung proximal and distal progenitors (Fig. 3G), with their shifts in strength perfectly reflecting their developmental succession [55]. In another example, when human pancreatic β cells were treated with FGF2 to induce de-differentiation into progenitors that did not have a ground-truth cell identity [56], we deduced that these developmentally upstream cells were of a fetal neuron identity (Fig. 3L). This trajectory has been suggested by previous studies on pancreatic development [57, 58]. Furthermore, across cases, we confirmed a strong resemblance of the cells at terminal differentiation points to fetal tissues rather than their mature counterparts (Fig. 3B–G), an intriguing phenomenon that has received extensive observations in diverse tissue contexts, including in brain [59,60,61], lung [62, 63], liver [64, 65], kidney [66, 67], and heart [68, 69]. Therefore, these results demonstrated that (i) our decomposition strategy was highly sensitive to the dynamics of lineage transformation and (ii) it can quantitatively resolve the uncertainty of cell states within a transiently heterogeneous phase, which is a hallmark of most physiological and disease processes, including the initiation and progression of cancer.
Defining a developmental-status aware, cell state panorama of human cancersGiven the biologically meaningful cell-state transition results observed in in vitro experiments, we next applied our decomposition strategy to characterize the cell states of patient cancer samples from The Cancer Genome Atlas (TCGA) [4]. We found dramatic heterogeneity of adult and fetal-like cell states within and between different cancer types (Additional file 3: Fig. S2). Importantly, the decomposed cell state proportions showed significant correlations with well-annotated clinical and molecular features. In the brain tumor cohort (pan-glioma, Glioblastoma/Lower Grade Glioma, GBM/LGG), we discovered a significant depletion of adult neurons and an enrichment of multiple fetal cell types in the tumor samples compared to the normal adjacent to tumor (NAT) samples (Fig. 4A). Among the normally abundant adult cell types that also exist in tumor samples, the presence of astrocytes and oligodendrocytes significantly correlated with the corresponding tumor histology (Fig. 4B), indicating a good agreement with the known cells of origin. The GBM samples have been previously assigned to four molecular subtypes that largely reflect their cell states, namely, proneural, classical, neural, and mesenchymal [70, 71]. Consistent with the observation that proneural GBM tumors are enriched in neurogenesis processes, we found a significant over-representation of fetal radial glia cells, a major neuronal progenitor [72], in that subtype (Fig. 4C). Gliomas are known to be highly infiltrated by microglia/macrophages [73, 74]. Accordingly, our decomposition results not only confirmed the richness of microglia in GBM samples but also revealed an increasing trend of its content with disease progression (Fig. 4D). Additionally, we observed that fetal radial glia cells were significantly favorable in IDH1 mutant tumors (Fig. 4E), reminiscent of the role of this single most pervasive driver event in GBM/LGG in promoting an undifferentiated cell state [75].
Fig. 4
Cell state decomposition map of TCGA tumor samples. A, F, and K Heatmaps showing unsupervised clustering of inferred cell fraction profiles of tumors and adjacent normal samples in the pan-brain, GBM/LGG (A), pan-kidney, KIRC/KIRP/KICH (F), and pan-lung, LUAD/LUSC (K) cohorts. Feature tracks presented on the top of the heatmaps show mutation status, tumor grade/stage, histology types, and smoking status. B–E Violin plots showing differential cell fractions across three LGG histological subtypes (B), four established GBM molecular subtypes (C), three brain tumor grades (D), and LGG samples stratified by IDH1 mutation status (E). G–J Violin plots showing differential cell fractions across three kidney histological subtypes (G), KIRC samples stratified by VHL mutation status (H), KIRP samples stratified by MET amplification status (I), and four KIRC tumor stages (J). L–O Violin plots showing differential cell fractions across two lung histological subtypes (L), LUSC samples stratified by TP53 mutation status (M), LUAD samples stratified by SOX2 amplification status (N), and LUAD samples stratified by smoking status (O). A two-sided Mann–Whitney U-test or a one-way ANOVA (Kruskal–Wallis) test was used to calculate the p-value
In the kidney cancer cohort (pan-kidney, clear cell renal cell carcinoma/chromophobe renal cell carcinoma/papillary renal cell carcinoma, KIRC/KICH/KIRP), we observed a dramatic shift of cell identities between NAT and tumor samples as well as among tumors of different histological types (Fig. 4F). Assessing the cell origins of the three kidney cancer types, we found that KICH showed significantly higher similarity to normal adult intercalated cells, while KIRC was enriched in proximal tubule cells and glomerular endothelial cells (Fig. 4F, G), both consistent with previous studies [76, 77]. However, we detected a significant over-representation of Henle’s loop cells in KIRP, which deviated from the claim in the above-mentioned studies that KIRP and KIRC shared common cells of origin. Since these studies relied on gene expression correlations of tumor samples with normal bulk samples to infer the cells of origins, we suspected that the discrepancy could be due to improved resolution of our decomposition results using single-cell signatures. We then investigated the impacts of two major pan-kidney driver events [78, 79] on the cell state of the cancer types: VHL somatic mutations in KIRC contributed significantly to a cell state that favored S-shaped body medial cell (Fig. 4H), a known precursor for Henle’s loop and the distal tubule [80]; MET amplification was associated strongly with the accumulation of distal tubule progenitor cells in KIRP (Fig. 4I). Moreover, we detected a significant positive correlation between the tumor stage and the infiltration of dendritic cells in KIRC (Fig. 4J), suggesting a trend of increasing immunogenicity during the progression.
In the lung cancer cohort (pan-lung, lung adenocarcinoma/lung squamous carcinoma, LUAD/LUSC), in contrast to the observation that the NAT samples of the two diseases were uniformly enriched in AT2 cells, macrophages and fibroblasts, we uncovered a striking divergence of the cell states between LUAD and LUSC (Fig. 4K). Specifically, LUAD was characterized by a significantly higher fraction of type I alveolar cells (AT1), while LUSC was enriched in distal progenitor cells (Fig. 4L), a cell type known to be a multipotent progenitor population in the lung [81]. The abundance of such cells was positively correlated with TP53 mutations and SOX2 amplification in LUSC and LUAD, respectively (Fig. 4M, N). These results suggest a close relationship between early lung speciation and lung cancer initiation in terms of the cell of origin, as suggested in a recent study [82]. Interestingly, we observed extensive macrophage infiltration in the LUAD cohort associated with the patient’s smoking status (Fig. 4O), a connection independently reported before in normal lungs [83, 84]. Collectively, our decomposition strategy represents a powerful approach to understanding the cells of origins and cell states of clinical tumor samples.
Quantifying cancer stemness based on a fetalness indexThe above analyses on individual cancers showed a consistent trend for tumor samples to be overall significantly enriched in tissue-specific fetal cell programs (Fig. 4A, F, and K). This observation suggested that through a process of reverse evolution, cancer cells yielded a more fetal-like cell state. To confirm this observation on a larger scale, we combined UMAP- projection-based visualization and PCA-based linear distance computing to explore the global topology of decomposed cell states of normal and tumor samples (TCGA/GTEx/EvoDevo), together with the original HCL single-cell reference set (see the “Methods” section). We found consistent proximity of tumor samples with either real fetal references or inferred fetal samples, compared to normal samples, across 12 cancer types (Fig. 5A and Additional file 3: Fig. S3A–S3G). Therefore, we reasoned that the aggregated fetal cell programs within a sample could be an informative index that can capture the evolutionary distance of a tumor cell state on a trajectory pointed towards a fetal-like state. Interestingly, when decomposing the TCGA samples using a mouse-based developmental cell-type reference, namely Mouse Cell Atlas (MCA) [18], we obtained similar results as in human-based decomposition (Additional file 3: Fig. S4A–S4I), suggesting a species-conserved adult-fetal transcriptome separation that mirrors cancer cell states. Using this index, we found that tumors across all the 12 cancer types had significantly higher fetalness than matched NAT samples and normal samples (Fig. 5B). We further confirmed these results based on non-TCGA cohorts of tumors and matched NAT samples (Additional file 3: Fig. S5A–S5L).
Fig. 5
留言 (0)