
記住我
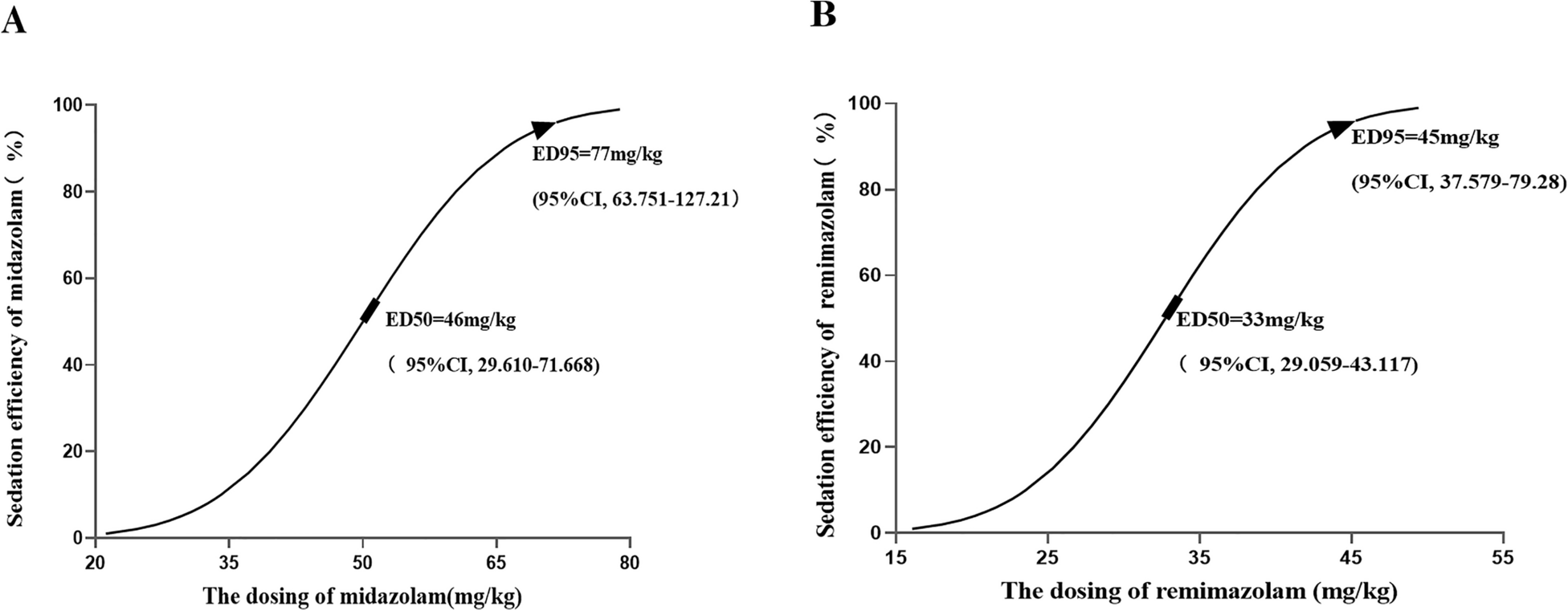


Type 2 diabetes (T2D) is a chronic condition characterized by insulin resistance and the progressive dysfunction of pancreatic β-cells, leading to persistent hyperglycemia. In this context, macrophages, key cells of the innate immune system, play a fundamental role in responding to inflammatory stimuli and contributing to metabolic stress in pancreatic tissues [9]. The interaction between elevated circulating glucose levels and the secretion of pro-inflammatory cytokines, such as IL-1β, induces an inflammatory response that exacerbates pancreatic islet dysfunction and promotes the polarization of macrophages toward a pro-inflammatory profile [51].
Insulin resistance, a central factor in the development of T2D, is intensified by the activation of inflammatory pathways in macrophages, such as the JNK and IKKβ pathways. These pathways activate serine/threonine enzymes that phosphorylate IRS-1, impairing insulin signaling [31]. Additionally, these pathways stimulate nuclear factor-κB (NF-κB), which increases the production of pro-inflammatory cytokines and nitric oxide (NO), elements that not only perpetuate inflammation but also aggravate insulin resistance [32, 40].
Under normal conditions, pro-regenerative macrophages predominantly rely on fatty acid metabolism and arginase-1 to maintain tissue homeostasis, reducing NO production [4]. However, the altered metabolic environment of T2D shifts macrophages to a pro-inflammatory phenotype, characterized by an increased reliance on glycolysis and NO synthesis via iNOS, fueling an inflammatory cycle that intensifies metabolic dysfunction [6, 45].
Metabolic variables related to insulin resistance, which contribute to the worsening of metaflammation, have been characterized using various methodological models, such as the use of mice, specifically in a lipodystrophic diabetic mouse model [53]. Additionally, in functional in vitro angiogenesis assays, the consequences of hyperglycemia were evaluated by simulating diabetic conditions [57]. In vivo models, especially in rodents, are widely used to analyze molecular, mechanistic, and phenotypic changes in transgenerational metabolic and cardiovascular programming. These models offer considerable advantages, such as short gestational periods and more offspring, allowing for the analysis of complex transgenerational mechanisms [8]. Therefore, these models enable a detailed analysis of underlying mechanisms, ensuring a more applied understanding of the consequences that T2D can have on immunometabolic interactions.
Thus, the objective of this article is to gather and analyze crucial information from the literature on the metabolic reprogramming of macrophages in the context of T2D, focusing on the main regulatory pathways that modulate macrophage polarization. Additionally, this review aims to discuss the impact that the diabetic context has on the innate immune system, specifically on macrophages, promoting a subclinical and persistent inflammatory alteration that directly affects the quality of life of millions of individuals. The review is organized into four main sections, exploring the impact of hyperglycemia on biochemical, metabolic, phenotypic, and epigenetic aspects, with the goal of expanding knowledge on the immunometabolic aspect of T2D.
The role of macrophages in T2DTissue-resident macrophages are long-lived cells derived from embryonic precursor cells, maintained through local proliferation [5]. These macrophages retain past immunological memories through trained immunity, which can alter their identity and impact acquired dysfunction over the years [5]. The inflammatory responses of macrophages are facilitated by changes in their cellular metabolism, where cells producing inflammatory mediators are induced to glycolysis, while inflammation is balanced by the stimulation of tissue repair [37]. Single-cell RNA sequencing studies have demonstrated that classifying macrophages as “M1” or “M2” does not adequately reflect the high heterogeneity present in tissues in vivo [36]. Instead of distinct populations, M1 and M2 signatures do not necessarily exclude each other and often coexist, depending on the balance between activating and inhibitory signals and the tissue environment [25]. For this reason, this review focused on highlighting predominantly pro- or anti-inflammatory phenotypes.
In Type 2 Diabetes (T2D), macrophages are reprogrammed to facilitate and exacerbate the inflammatory response when continuously exposed to pro-inflammatory stimuli and metabolic changes in tissues and organs, leading to metaflammation [26, 37]. This condition is characterized by chronic low-grade inflammation, which can result in various complications and comorbidities. The exacerbated inflammation is evidenced by markers, such as elevated levels of C-reactive protein (CRP), increased expression of pro-inflammatory cytokines, and a higher white blood cell count, notably a 20% increase in the monocyte population compared to the global population, which can be readily recruited to tissues under inflammatory conditions [5]. Although other immune cells participate in metaflammation in T2D, macrophages are the primary effector cells leading to reduced insulin sensitivity [36].
A study conducted by Valtierra-Alvarado and colleagues demonstrated that obese and diabetic patients exhibit a reduction in classical monocytes, which are associated with antimicrobial function, while there is an increase in monocytes that predominantly perform antigen processing and presentation. This suggests a close relationship between the reduction of classical monocytes in poorly controlled T2D and the susceptibility to multiple infections as a comorbidity of the disease. However, the same study showed that inadequate glycemic control negatively affects the expression of HLA-DR, a surface marker of antigen-presenting cells (APCs), such as macrophages and monocytes, as well as CD86, a co-stimulatory molecule. HLA-DR is responsible for presenting peptides to T lymphocytes, which interact with CD86 to promote T-cell activation and differentiation. This suggests that antigen presentation is also impaired in T2D, leading the patient to an immunosuppressed state [44].
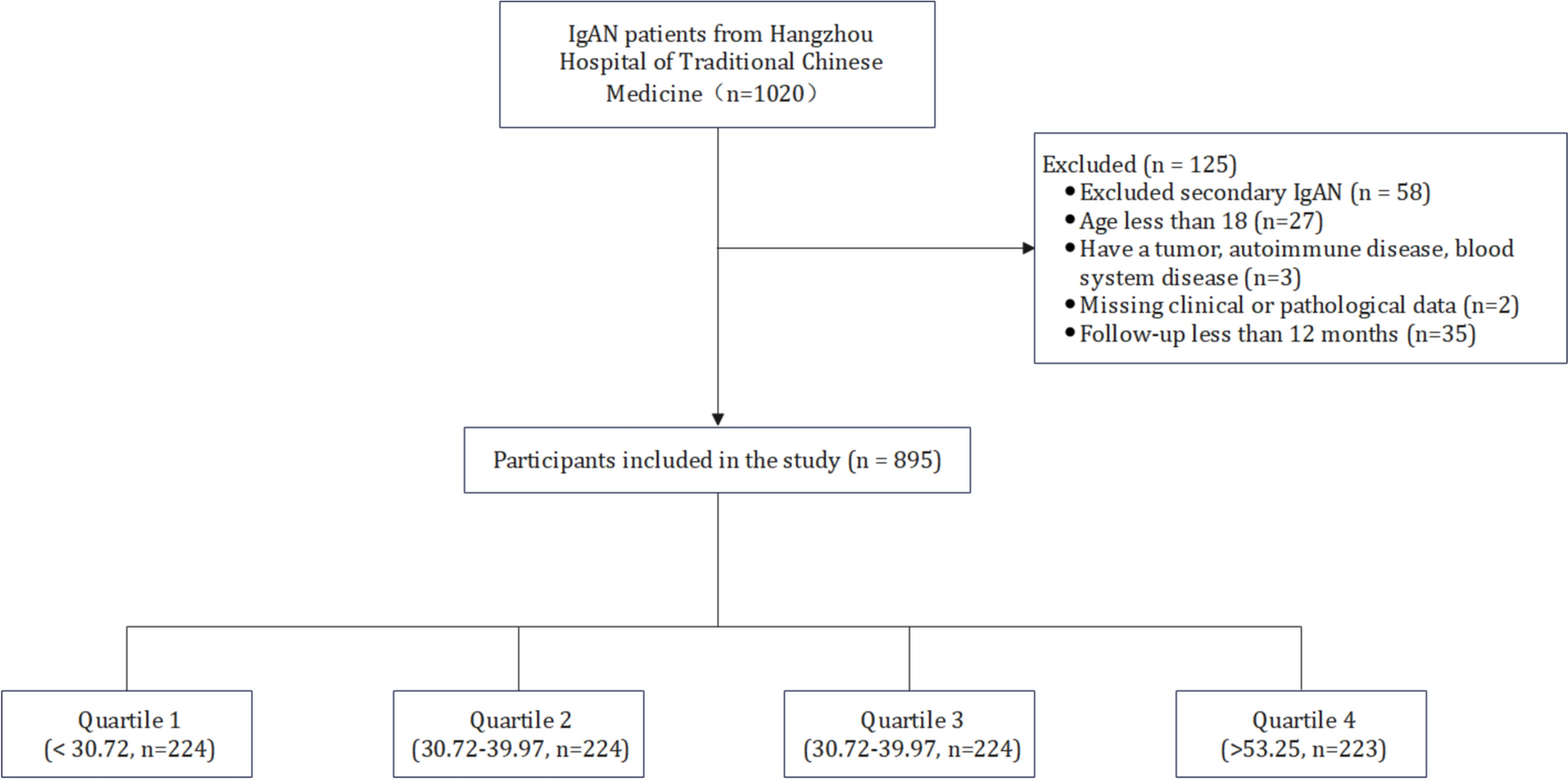
In addition to playing a crucial role as APCs, macrophages are essential in glycemic control by contributing to insulin secretion by β-cells [55]. However, in T2D, macrophages are reprogrammed to impair this process and are the main contributors to inflammation in pancreatic islets [55]. Studies in mice have demonstrated that the inhibitory effect of macrophages on glucose-stimulated insulin secretion (GSIS) depends on direct contact with β-cells through open cytoplasmic channels, through which a greater number of intact insulin secretory vesicles pass from β-cells to macrophages. Other factors also contribute to the reduction of GSIS and the increase in β-cell dysfunction [55]. The decrease in GSIS precedes an adaptive expansion of β-cells in the pre-diabetic phase, mediated by the interaction of platelet-derived growth factor (PDGF), produced by macrophages, with its receptor (PDGFR) on β-cells [55] (Fig. 1). This occurs, because hyperglycemia increases as insulin resistance worsens, until prolonged insulin production, glucolipotoxicity, oxidative stress, inflammation, and β-cell dedifferentiation result in functional decline and apoptosis [5] (Fig. 1; Table 1).
Fig. 1
Schematic representation of the proliferation of pro-inflammatory macrophages and β-cells in the diabetic pancreatic islet and the signaling mechanisms and interactions between pro-inflammatory macrophages and β-cells. Excess free fatty acids, glucose, and ATP stimulate macrophage proliferation, which in turn, by secreting pro-inflammatory cytokines, induces endoplasmic reticulum (ER) stress, production of pro-apoptotic proteins, and activation of the NF-Kβ pathway, which inhibits cell differentiation genes and insulin secretion. Additionally, interactions via the cytoplasmic channel (CT) affecting glucose-dependent insulin secretion and the PDGF-PDGFR signaling, stimulating β-cell proliferation in the islet are represented
Table 1 Role of macrophages in type 2 diabetes (T2D) by tissuePatients with T2D exhibit elevated levels of CD68+ macrophages in the islets, primarily due to the local proliferation of resident macrophages, stimulated by high levels of glucose and free fatty acids (FFAs) [5, 47, 55]. Some of these signals are secreted by the stressed β-cells themselves, contributing to the infiltration and activation of more macrophages through the release of chemokines and ATP [55] (Fig. 3). ATP-stimulated macrophages increase the secretion of pro-inflammatory cytokines, such as IL-1β, TNF-α, and INF-γ, inducing the activation of pro-apoptotic proteins, endoplasmic reticulum stress, and a consequent reduction in GSIS, in addition to activating the NF-Kβ pathway, which acts to reduce the expression of cell differentiation genes and insulin secretion in β-cells [55] (Fig. 3; Table 1).
The prolonged activation of the NF-Kβ pathway in adipocytes stressed by pro-inflammatory macrophages is also responsible for increasing the expression of non-canonical kinases, which attenuate β-adrenergic signaling in adipose tissue by participating in the phosphorylation of catecholamines, reducing energy expenditure and consequently increasing the accumulation of FFAs [35]. Additionally, adipose tissue macrophages (ATMs) have a greater capacity to degrade catecholamines in T2D, resulting in a reduced response to thermal stress, once again contributing to lower energy expenditure [22] (Table 1).
Like pancreatic islets, white adipose tissue (WAT) undergoes a significant increase in the number of macrophages in T2D, which can reach up to 40% more than in a healthy individual [5]. In diabetic individuals, ATMs adopt a metabolic activation state (MMe), characterized by a pro-inflammatory phenotype along with increased lysosomal activity and survival, aiming to phagocytize necrotic adipocytes, which elevates lipolysis and FFA levels [37, 44]. The accumulation of triglycerides in T2D increases the volume and number of adipocytes, such that the capillary network, orchestrated by macrophages, cannot keep pace, leading to hypoxia and adipose tissue stress [26]. This stimulates inflammation, lipotoxicity, and, eventually, macrophage infiltration, which forms crown-like structures around damaged adipocytes [26].
The excess of FFAs derived from adipose tissue and diet in T2D contributes to the pro-inflammatory activation of hepatic macrophages, known as Kupffer cells (KCs), leading to increased pro-inflammatory cytokines and the activation of the hepatic NF-Kβ pathway, contributing to the development of non-alcoholic steatohepatitis (NASH), a pathology commonly associated with T2D [5, 47]. Hepatocytes affected in NASH further activate KCs and recruit more monocytes to the liver, increasing levels of cytokines, and reactive oxygen species (ROS), which cyclically exacerbate NASH and insulin resistance [47] (Table 1).
Studies have also shown that levels of Escherichia coli, a significant source of lipopolysaccharides (LPS), are higher in patients with T2D [47]. LPS is recognized by Toll-like receptors on macrophages, leading again to the activation of the NF-Kβ pathway, which is widely associated with intestinal integrity and permeability, as well as insulin resistance [47]. Due to increased intestinal permeability in T2D, microbial products leak through the portal circulation, exacerbating inflammation and insulin resistance in the liver [47]. Moreover, a high-fat diet induces intestinal epithelial cells to produce monocyte chemotactic proteins, exacerbating chronic inflammation in the intestinal lamina propria and perpetuating endotoxemia [47] (Table 1).
Biochemical alterations within macrophages in T2DThe metabolic status of macrophages plays crucial roles in the development of immune responses. The population of pro-inflammatory macrophages is highly dependent on the glycolytic pathway, while the population of pro-resolutive ones utilizes the fatty acid oxidation pathway [21]. In the context of T2D, characterized by chronic hyperglycemia, driven mainly by insulin resistance, there is a high rate of glucose auto-oxidation, resulting in the synthesis and accumulation of reactive oxygen and nitrogen species in the body [27]. Additionally, the excessive available glucose is converted into fatty acids and stored as lipids, particularly triacylglycerols intracellularly [28].
Adipocyte hypertrophy, hypoxia, and increased cell death due to lipid accumulation contribute to the secretion of pro-inflammatory molecules, such as TNFα, IL-1β, IL-6, and IL-8 [42], which promote the recruitment of monocytes that tend to differentiate into pro-inflammatory macrophages [20]. These pro-inflammatory stimuli, in turn, activate JNK and IKKβ pathways, promoting insulin resistance by phosphorylating serine residues on IRS-1 and by transcriptional activation of nuclear factor-κB (NF-κB), respectively [40].
Phosphorylation sites of the JNK pathway include Ser312—in humans, and in mice, the site is called Ser307—as the primary site for protection by restriction of the phosphotyrosine-binding domain [1] and Ser302, which has also been shown to mediate the disruption of IRS-1 signaling. It is also important to highlight the importance of phosphorylation of other additional sites for this disruption to occur [49].
Once activated, serine/threonine enzymes act on NF-κB transcription, which, upon interaction with the cell nucleus, stimulates the production of cytokines, pro-inflammatory mediators, and iNOS, responsible for NO production. NO, upon interaction with the insulin receptor, leads to insulin signaling resistance [32], therefore contribution to the worsening of the clinical condition. Activation of MAP-kinase signaling pathways promotes ET-1 secretion, activates cation pumps, and increases the expression of VCAM-1 and E-selectin [39]. These molecules enhance monocyte adhesion to endothelial cells, where they differentiate into macrophages and produce inflammatory molecules, further promoting local inflammation [40].
Metabolism in the context of T2DThe regulation of different metabolic profiles in macrophages has profound implications for the immune response, being closely linked to the development of diabetic and inflammatory conditions [13]. As evidenced in the previous section, in homeostatic situations, macrophages meet their energy demands predominantly through oxidative phosphorylation (OXPHOS) [3]. During this state, glucose is absorbed by specific receptors present on the cell membrane of macrophages, such as GLUT, and directed toward glycolysis, generating pyruvate through pyruvate dehydrogenase (PDH), which is then internalized into the mitochondria and utilized in the tricarboxylic acid (TCA) cycle (Fig. 2) [3, 12, 11]. This efficient process produces ATP through OXPHOS, taking advantage of the presence of oxygen.
Fig. 2
Disruption of the citric acid (TCA) cycle to citrate and succinate causes significant modulations in the glycolytic pathway. The increase in hexokinase (HK) promotes the increase in fructose-6-phosphate (F6P), which activates the pentose phosphate pathway (PPF), resulting in the production of NADPH and reactive oxygen species (ROS). The action of HIF-1α inhibits pyruvate dehydrogenase, favoring the conversion of pyruvate into lactate by lactate dehydrogenase. The increase in succinate, due to the anaplerotic activity of glutamine, affects the electron transport chain, particularly complex II, inducing reverse electron transport (RET) and generating more ROS, which increases the expression of IL-1β, a cytokine pro-inflammatory
On the other hand, the microenvironment presented in T2D directs the macrophage profile toward a pro-inflammatory profile. These changes in macrophage metabolism under glucose excess exhibit similarities to the Warburg effect [3, 32, 48]. The Warburg effect, a phenomenon first observed in 1924 by Otto H. Warburg when studying malignant tumors, describes the increase in glucose uptake by cells and its conversion into lactate even in the presence of oxygen, an environment in which oxidative phosphorylation would be the most efficient, proposed as an adaptation mechanism to support the cell’s biosynthetic needs, characterizing the term aerobic glycolysis [46]. This deviation contributes some advantages to the cell, promoting the excess production of carbon sources, which are subsequently used to generate other macromolecules, such as nucleotides, lipids, and proteins [24, 54]. In addition, it also allows the generation of ATP, which when in the Warburg phenotype is less efficient compared to mitochondrial respiration, but is accelerated, since the rate of glucose metabolism through aerobic glycolysis allows the production of lactate 10–100 times faster than complete oxidation in the mitochondria [24, 54]. Studies and calculations indicate that cells with a higher rate but lower yield in ATP production may have a selective advantage when competing in microenvironments with shared and limited energy resources [24, 54]. The molecular mechanisms involved in the Warburg effect have not yet been clearly established, but studies suggest that the hypoxia-induced transcription factor (HIF) is important [19].
As in the Warburg effect, this pro-inflammatory metabolic profile in T2D is characterized by increased glucose uptake, resulting from the overexpression of glucose transporter 1 (GLUT1), which is characterized as the main glucose transporter in macrophages, increasing glucose uptake independently of insulin signaling via IRS-1 [12, 11, 30]. This process is mediated by the activation of HIF-1α, which is stabilized and induced by the combination of increased glycolysis and the release of pro-inflammatory cytokines resulting from metabolic stress [30]. HIF-1α also induces the expression of other genes that encode enzymes with glycolytic function, such as glucose-6-phosphate dehydrogenase and hexokinase (HK) [12,
留言 (0)