
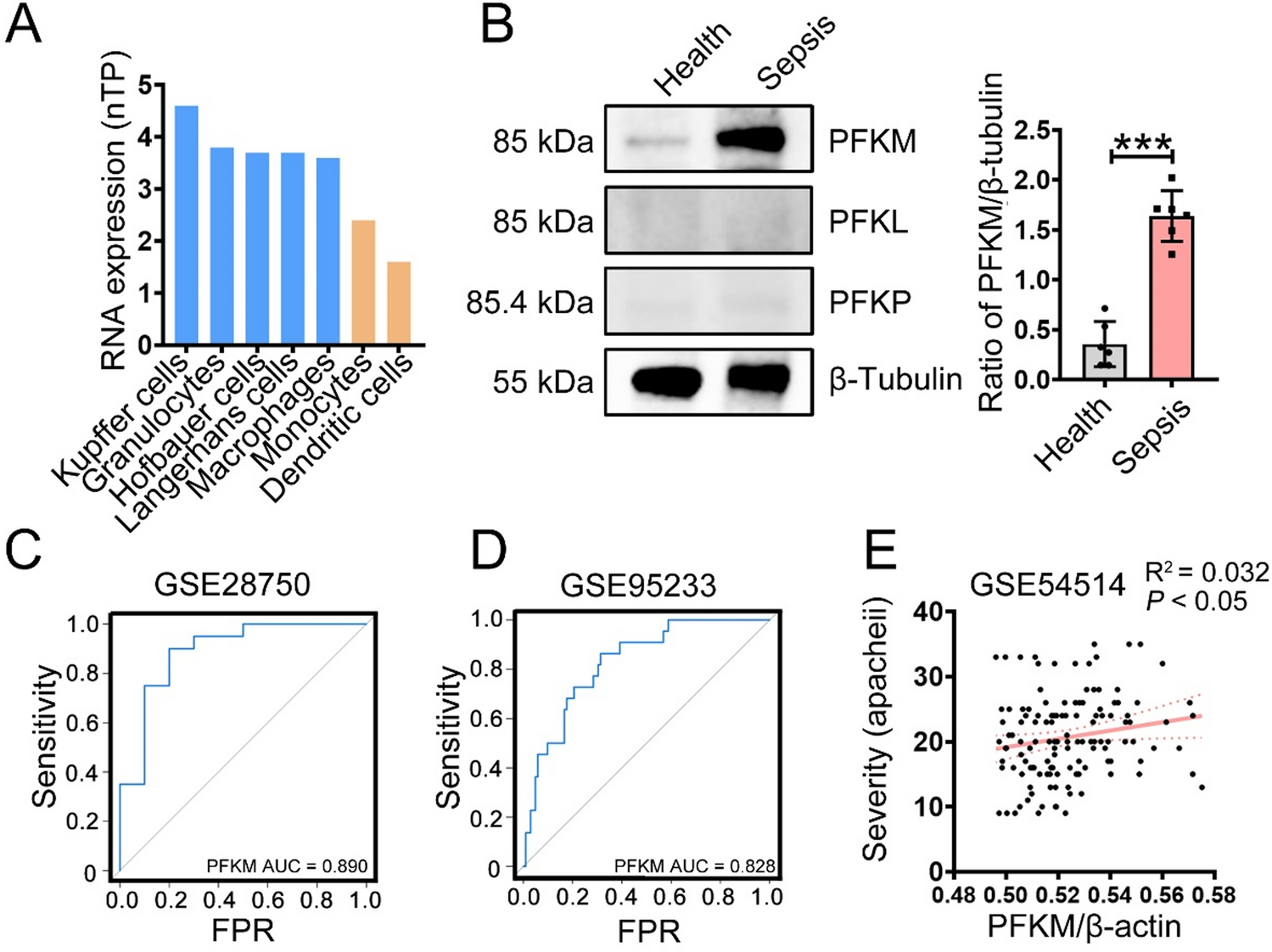
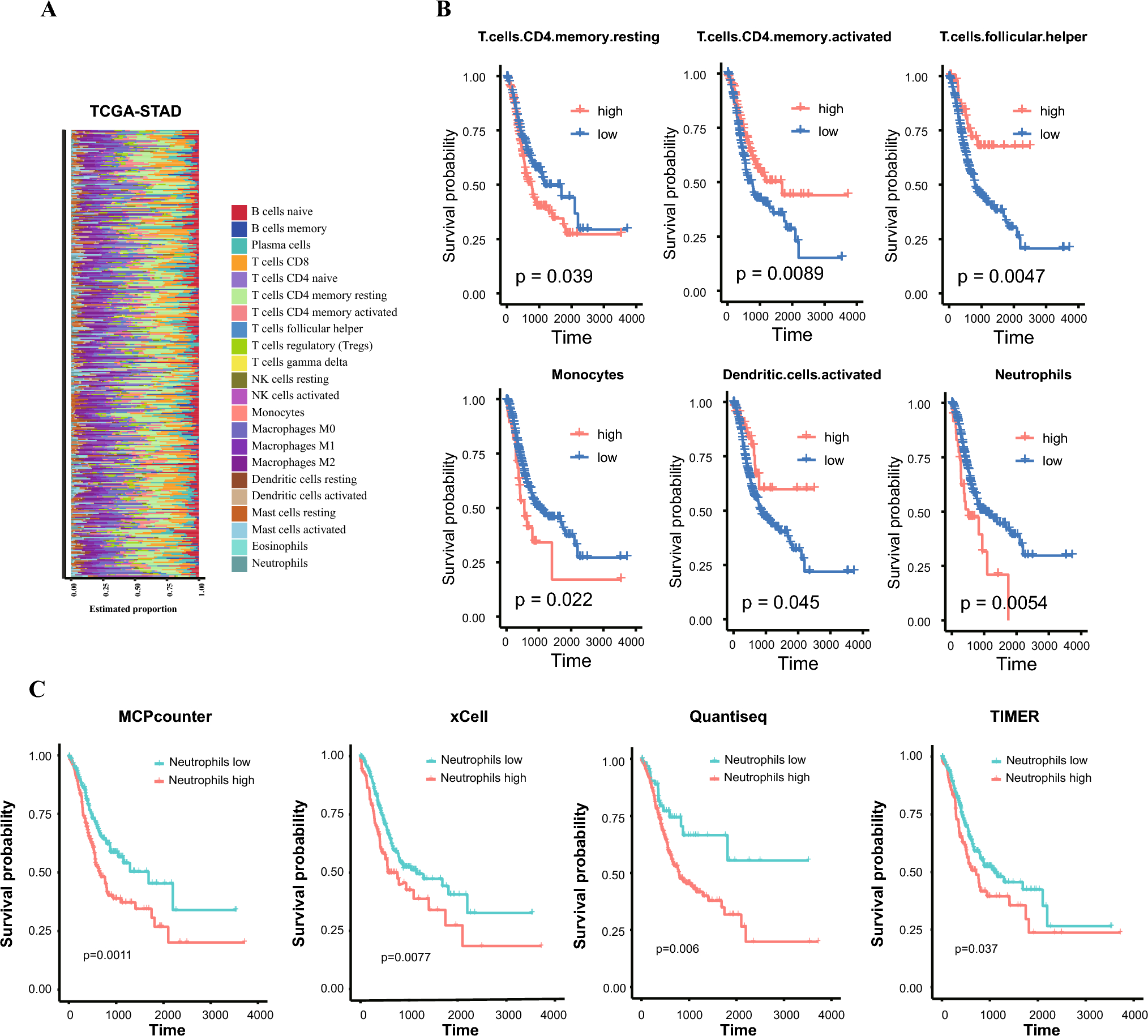
記住我




We have previously shown that Cx36 mutants that lack the final four amino acids, which constitute the PDZ binding motif (PBM), exhibit a trafficking defect that causes the connexin to accumulate in the ER. As a result of this retention, Cx36 channels inside the ER are allowed to dock prematurely leading to the formation of gap junction like aggregates that reshape the ER membrane into concentric whorls (Fig. 1A). These structures can be found in the cytosol (illustrated in the cartoon in yellow, Fig. 1A) and around the nucleus (illustrated in magenta, Fig. 1A) and can be recognized via their characteristic toroid shape (whorls) in immunofluorescent imaging [19]. In HEK293T cells transiently expressing wild type Cx36, the connexin forms perinuclear structures that colocalized with the SNARE proteins Syntaxin5 and Sec22, localizing at the ER-Golgi interface (Fig. 1A). The association of Cx36 with both of these proteins implies, that the overexpressed connexin is trafficked via components of the secretory machinery present in HEK293T cells. Interestingly, although we observed functional gap junction formation, we found that Cx36 did not colocalize with endogenous ZO-1 in transfected HEK293T cells, suggesting that trafficking of connexons in the early secretory pathway is independent of ZO-1 in this context (Fig. 1A).
Fig. 1
The C-terminal “YV motif” of Cx36 is necessary for ER export. A Left: Cartoon illustrating the ER retention mechanism in C-terminal Cx36 mutants. Right: Colocalization of Cx36 with Stx5 and Sec22 in perinuclear regions. Cx36 in transfected HEK293T cells does not associate with endogenous ZO-1. B, C Insertions or deletions of amino acids at the very C-terminus result in ER retention. Deletions of amino acids N-terminal from the YV motif do not lead to ER retention and whorl formation. Scale in A and B: 10 µm. Values are shown as mean ± standard error of mean. Significance was tested using a one way ANOVA. Cx36 mutants in which the very C-terminus was modified (mutation surrounding the YV sequence) show a significant increase in whorls and nucleus associated vesicles. P < 0.01. D GFP trap pull-down of PDZ1 (ZO-1) co-expressed with different Cx36 mutants. Asterisk marks the Cx36 dimer band
In the course of this study we used the PBM-deficient Cx36 whorl phenotype as a marker for ER retention to identify factors that control the functional ER export of Cx36. In a first set of experiments we asked whether the PBM alone, certain amino acids within the motif, or additional residues in the C-terminal tail mediate the ER export of Cx36. To identify a minimal motif we generated several Cx36 mutants that lack portions of the C-terminal tail (mutant sequences in Fig. 1A) and expressed these constructs in HEK293T cells. For each mutant, we determined the number of ER whorls in the cytosol (yellow bar graph) and vesicles that are surrounding the nucleus (magenta bar graph), as both of these structures are increasingly formed in HEK293T cells that express ER retained Cx36 mutants. Our mutagenesis screen revealed a significant increase in the number of ER whorls and nucleus associated puncta for Cx36 mutants that affected the very C-terminus comprising the amino acids tyrosine (Y) and valine (V) (Fig. 1B, C). Attaching a FLAG tag to the C-terminus caused similar trafficking defects, suggesting that a functional carboxyl group of the valine residue is part of the export signal. This is consistent with the trafficking defects that have been described for the Cx36-EGFP construct [15, 16]. When we deleted additional amino acids upstream from the YV motif and PDZ binding domain or directly within the C-terminal tail (Cx36Δ294-298, Cx36Δ299-303) we found no signs of ER retention. When we replaced the PDZ binding motif of Cx36 with an artificial sequence (GLGF) that matches the criteria for a class II PDZ binding motif (Φ-X-Φ-COOH), we observed an increase in the number of nucleus associated puncta but no signs of ER whorl formation.
Our mutagenesis screen demonstrated that deletion of tyrosine 320 or valine 321 is sufficient to reproduce the ER export deficit we described for the S318/ter mutant. At this point, however, it is unclear if the ER export defect arises from the loss of a separate export signal or from the disruption of PDZ domain mediated interactions. To address this issue, we tested the ability of each PBM mutant to bind the PDZ1 domain of ZO-1 (Fig. 1D). We co-expressed a venus-tagged PDZ1 domain with each Cx36 mutant in HEK293T and performed a GFP trap pulldown. Of the constructs tested, those which showed the whorl phenotype were unable to bind, and only wild type Cx36 and the Cx36Δ313-319 bound PDZ1. Additionally, we found that the Cx36Δ313-319 mutant showed a massive increase in PDZ binding, although it is lacking the first two amino acids of the PBM. This drastic effect is likely due to the repositioning of the YV motif next to the upstream sequence RT creating a high affinity PDZ ligand with following sequence: RTYV. Taken together, we find that Cx36 mutant that showed ER retention (whorls) also failed to bind PDZ1, raising the question whether functional ER export requires the entire PBM “SAYV” or the “YV” motif.
Functional ER export of Cx36 requires the cargo receptors Sec24A and BOur data have shown that an intact C-terminus of Cx36 determines the functional ER export of Cx36 (the term functional is used intentionally to distinguish COPII mediated export from whorl formation, which also displays features of an export mechanism). The release of newly synthesized proteins from the ER in eukaryotic cells is regulated by the COPII complex. This conserved release machinery requires the assembly of cytosolic coat proteins that deform the ER membrane at ER exit sites into secretory vesicles containing the cargo protein [22]. Within the COPII complex cargo recognition is controlled by Sec24 isoforms, a family of adapter molecules that connect the cargo protein to the COPII coat via interactions with certain export signals in its cytoplasmic domains [23]. As the COPII complex functions as an almost universal export route for most proteins, we reasoned that the trafficking deficient Cx36 mutants may fail to interact with the Sec24 cargo receptors preventing them from entering the COPII vesicle and resulting in ER retention. To address this hypothesis, we first tested if inhibition of the COPII complex reproduces the phenotype we observed for trafficking deficient Cx36 mutants. The assembly of the COPII coat is initiated by the small G protein Sar1. We co-expressed the dominant negative Sar1 (T39N)-GFP mutant, which serves as a competitive inhibitor of endogenous Sar1 [24], with wild type Cx36 to block COPII complex formation and analyzed the intracellular distribution of the connexin. Sar1(T39N)-GFP co-transfection caused an accumulation of Cx36 into dense perinuclear vesicles (magnified inset) and the formation of smaller aggregates surrounding the nucleus (white arrows) (Fig. 2A). Especially the nucleus associated puncta (white arrows) were reminiscent of the trafficking defect we have described for Cx36 mutants lacking the PDZ binding motif. Interestingly, we observed that co-transfection of the COPII coat protein Sec23 disrupted the transport of wild type Cx36 in a similar way as removal of the PDZ binding motif. Sec23-GFP transfected cells formed the characteristic connexin whorls (magnified inset) and smaller aggregates surrounding the nucleus (white arrows) (Fig. 2A). Although Sec23-GFP doesn’t function as a COPII inhibitor, it is likely that the overexpressed coat protein depleted binding sites for the endogenous Sec23 within the COPII complex and thereby prevented the release of newly synthesized Cx36 channels. When we co-expressed Sec61B-GFP, an ER membrane protein that does not belong to the COPII complex, we did not see any signs of ER retention, confirming that ER export of Cx36 is COPII dependent.
Fig. 2
Cx36 requires COPII cargo receptors for ER export. A Overexpression of Sar1T39N-GFP and Sec23A-GFP lead to accumulation of Cx36 around the nucleus as previously described for the Cx36/S318ter mutant. B Western blots confirming siRNA mediated depletion of secretory pathway proteins. C Confocal scans of Cx36 in siRNA treated cells. Knock-down of specific Sec24 isoforms has distinct effects on the distribution of Cx36. Combined knock-down of Sec24A and B reproduces the trafficking defect observed for the Cx36/S318ter. Knock-down of Sec24C, on the contrary, leads to accumulation of Cx36 in perinuclear structures. D Knock-down of ER-Golgi trafficking proteins Ergic3 and GOPC have no effect on Cx36 distribution, while knock-down of Syntaxin5 leads to accumulation of Cx36 in the perinuclear region. E Evaluation of different phenotypes observed in Sec24A/B or Sec24C depleted cells. Cx36/Stx5 PLA intensity was not significantly different. All p values were > 0.05. More ER whorls are formed in Sec24A/B depleted cells. Data are shown as mean ± standard error of mean. Significance was tested using a one way ANOVA. Scale: 10 µm
Next, we sought to identify the exact cargo receptor isoforms that bind to Cx36 in the COPII complex. We targeted all four Sec24 variants using siRNA and tested how silencing of each isoform influenced the intracellular distribution of Cx36. To validate the successful knock-down we tested the Sec24 protein level in cell lysates from each transfection (Fig. 2B). Knock-down of Sec24 A and B showed little to no effects on the distribution of Cx36. Only combined depletion of both isoforms led to the formation of the characteristic Cx36 whorls (magnified insets) and aggregates surrounding the nucleus (Fig. 2C), resembling the trafficking defect we have described for the Cx36S318Ter (Fig. 1B). Silencing of Sec24C or double knockdown of Sec24C and D, on the contrary, concentrated Cx36 into dense perinuclear structures.
Besides the Sec24 isoforms, we tested the impact of additional proteins that have been linked to intracellular trafficking events (Fig. 2D). Among them was Ergic3, a membrane protein that has been shown to control the ER to Golgi transition of Connexin43 and innexins (invertebrate GJ forming proteins [25]), suggesting a function as a universal export factor for metazoan gap junction proteins [26]. In our hands, however, depletion of Ergic3 did not cause any obvious signs of ER retention. Next, we targeted the Golgi specific PDZ protein GOPC (Golgi-associated PDZ and coiled-coil motif-containing protein). This protein appeared as an interesting candidate due the PDZ domain and role in the trafficking of claudin1 and 2, which carry C-terminal YV motifs identical to Cx36 [27]. Similar to the results for Ergic3, we did not observe any ER retention effects for Cx36 when we silenced GOPC. As a final candidate on our list we targeted Syntaxin5 (Stx5) a SNARE protein that fuses the membranes of ER export vesicles and the cis Golgi to allow the uptake of secreted cargos into the Golgi apparatus [28]. Interestingly, we found that depletion of Stx5 caused the formation of dense perinuclear clusters containing Cx36. As Stx5 depletion per se would not prevent the release of Cx36 from the ER, it is likely that these clusters represent secreted channels that accumulate around the Golgi apparatus.
We observed that silencing of Sec24A and B or Sec24C resulted in quite distinct intracellular distributions of Cx36. While depletion of Sec24A/B reproduced the ER retention phenotype for Cx36, knockdown of Sec24C, on the contrary, led to the formation of dense perinuclear clusters resembling the Golgi apparatus. We further evaluated these phenotypes and performed a proximity ligation assay to detect Cx36/Stx5 interactions. This strategy allowed us to label Cx36 channels that have transitioned into the Golgi apparatus since Stx5 is largely confined to the cis Golgi. In comparison to control siRNA treated cells, however, this assay only detected a subtle insignificant increase in Cx36/Stx5 in Sec24C depleted cells. The number of ER whorls in Sec24C depleted cells was significantly lower in comparison to Sec24A/B depleted cells, reflecting the lack of an ER retention phenotype when Sec24C was knocked down. In comparison to control siRNA treated cells, Sec24A/B depletion substantially increased the number of ER whorls (yellow bar graph) and vesicles surrounding the nucleus (Fig. 2E). While the increase in the frequency of nucleus associated vesicles in Sec24A/B depleted cells was highly significant compared to the control condition, the number of cytosolic whorls (yellow bar graph), however, was not. Though it appears, that Sec24A/B depletion reproduced only one of the two ER retention phenotypes, one has to consider that in comparison to simple overexpression experiments (Fig. 1) a lower amount of the Cx36 expression vector (333 ng) had to be co-transfected with Sec24 siRNAs to avoid cytotoxic effects. Since whorl formation heavily depends on the amount of Cx36 localizing in the ER, it is likely that our assessment of ER whorls in Sec24A/B depleted cells is an underestimate of the actual ER the retention effect.
To understand how Cx36 interacts with the COPII cargo receptors, we first analyzed the colocalization of the connexin with Sec24 A, B and C. Each of these receptors colocalized with Cx36 in perinuclear regions (Fig. 3A). Consistent with the knock down experiments, we observed a strong association of Cx36 with Sec24A and B, with around 60% of perinuclear Cx36 colocalizing with the two cargo receptors (Fig. 3B). A lower degree of colocalization was detectable for Sec24C. We next asked whether the cargo receptors Sec24B and C directly bind to Cx36 and co-transfected GFP-tagged Sec24 constructs with Cx36 into HEK293T cells to perform a pull-down assay (Fig. 3B). As a positive control we transfected the venus tagged PDZ1 domain of ZO-1 which showed substantial binding to Cx36. We observed no association with the connexin when the pull-down was performed with the Sec24B or C, suggesting that Cx36 does not directly interact with these cargo receptors. Another possibility to explain the lack of a detectable interaction is that Sec24 isoforms bind Cx36 with low affinity making co-precipitation experiments difficult. To bypass this issue and test a molecular association of Sec24B and Cx36 we performed a proximity ligation assay using the duo link PLA system. We transfected Cx36 into HEK293T cells and tested the PLA reactivity targeting Cx36 and endogenous Sec24B. Consistent with the double labeling experiments we observed strong PLA reactivity in perinuclear regions likely reflecting ER exit sites (Fig. 3C). A clear reduction in PLA labeling of these perinuclear regions was observed when the assay was performed with Cx36/S318Ter mutants lacking a functional C-terminus. Based on this observation we conclude that a molecular association with Sec24B involving a functional C-terminal tip of Cx36 is necessary to incorporate the connexin into COPII vesicles and transport it out of the ER.
Fig. 3
Cx36 associates with COPII cargo receptors. A Cx36 shows partial colocalization with the three different cargo receptors Sec24A, Sec24B and Sec24C. Line scans depict the overlap of intensity maxima for a selection region of interest. B Colocalization analysis illustrates the degree of colocalization for Cx36 and different COPII cargo receptors. The difference in Cx36 colocalization for each Sec24A and B was significant in comparison to Sec24C. Sec24A vs. Sec24C, P = 0.0032. Sec24B vs. Sec24C, P = 0.0024. Values are shown as mean ± standard error of mean. Significance was tested using a one way ANOVA. Immunoprecipitations fail to detect binding interactions of Cx36 and the COPII cargo receptors Sec24B and Sec24C. Venus-PDZ1-ZO-1 served as a positive control and showed a robust interaction with Cx36. C Proximity ligation assay was used to assess the molecular association of Cx36 and Sec24B. The Cx36 and Sec24 PLA reaction results in compact perinuclear signals that are significantly weaker in intensity when the assay is performed with Cx36/S318ter. Scale: 10 µm
Cx36 interacts with the Golgi stacking protein Grasp55To identify additional proteins acting within the secretory pathway to control Cx36 trafficking, we used a TurboID screening approach. TurboID is an engineered, bacterial enzyme that promiscuously biotinylates lysine residues on proteins within an ~ 10 nm radius of localization, subsequently allowing for easy isolation of the biotinylated proteins [29]. We first tried engineering TurboID directly onto Cx36, but found that insertion of various TurboID variants into Cx36 interfered with the functional transport of gap junction channels, irrespectively of the insertion site. We therefore turned to the recently developed BLITZ technique, in which a destabilized GFP-targeting nanobody carries TurboID to subcellular locations where proteins are tagged with GFP [19, 21] (Fig. 4A). Given that we found that disrupting the terminal “YV” motif resulted in ER export defects (Figs. 1–3), we co-expressed wildtype Cx36 and Cx36-EGFP in HEK293T cells and found that the tagged Connexin did not display the hallmarks of ER retention (Fig. 4A). The Cx36-EGFP construct is presumably being rescued by the wildtype Cx36 that is co-expressed, as has been observed in vivo in transgenic mice [15, 16]. We additionally co-expressed the destabilized GFP-targeting nanobody that carries TurboID (V5-TurboID-dGBP) and found that it colocalizes with Cx36-EGFP and results in substantial biotinylation at the plasma membrane (Fig. 4A arrows) and intracellular vesicles (Fig. 4A small arrow heads). This control experiment confirms that the TurboID construct is correctly targeting Cx36 without compromising the transport of gap junction channels. We isolated biotinylated proteins from Cx36-EGFP/Cx36/ V5-TurboID-dGBP co-transfected HEK293T cells, and as control, cells expressing only V5-TurboID-dGBP, and analyzed the samples via mass spectrometry. In Cx36-EGFP/Cx36 co-transfected samples we identified several secretory transport proteins including Sec24A/B, synergin gamma, and the Golgi reassembly stacking protein 55 (Grasp55) (Fig. 4A, B). Grasp55 was of particular interest for us given its function in unconventional protein secretion [30] and its two PDZ domains mediating interactions with proteins bearing C-terminal valines such as CD8 [31]. We first tested if Cx36 and Grasp55 interact and co-expressed GFP-tagged Grasp55 with Cx36. We isolated overexpressed Grasp55 from co-transfected HEK293T cells using GFP trap and co-precipitated substantial amounts of Cx36. When the pull-down was performed with a Grasp55 construct lacking the two PDZ domains or with a truncated mutant of Cx36 missing the PBM we observed no binding, indicating that Grasp55 interacts with Cx36 via its PDZ domain (Fig. 4C). We further observed that transfected Cx36 colocalized with endogenous Grasp55 in perinuclear structures resembling the Golgi apparatus (Fig. 4D). To demonstrate that Cx36 also interacts with endogenous Grasp55 in HEK293 we performed a proximity ligation assay and measured the intensity of perinuclear PLA signals in Cx36 transfected cells (Fig. 4E). In comparison to wild type Cx36 transfected conditions the Cx36S318ter mutant showed significantly weaker Cx36/Grasp55 PLA reactivity, suggesting that Cx36 binds to endogenous Grasp55 in a PDZ dependent manner. Besides Grasp55 we identified the protein tyrosine phosphatase non-receptor type 13 (PTPN13) in our BioID screen as an additional interactor of Cx36. PTPN13 consists of multiple of PDZ domains and functions as a tumor suppressor [32]. We have previously shown that perch Cx35, which has an identical PBM to Cx36, bound to the second PDZ domain of PTPN13 in a micro array screen [33]. Consistent with this finding, we observed extensive colocalization of transiently expressed Cx36 and endogenous PTPN13 at gap junctions in transfected HEK293T cells (Fig. 4F). We excluded PTPN13 as an additional secretory factor for Cx36 because the protein was mostly confined to gap junctions but rarely associated with intracellular vesicles. Thus, the only PDZ protein we identified in our BioID screen that could potentially affect trafficking of Cx36 in early secretory compartments is Grasp55.
Fig. 4
Cx36 and Grasp55 interact via PDZ/PBM binding and colocalize in the Golgi. A GFP directed proximity biotinylation was used to identify proteins regulating the transport of Cx36 in the secretory pathway. Immunostaining confirms correct targeting of V5-TurboID-dGBP and localized biotinylation. Heatmap summarizes the abundance of hits in the BioID screen. Among several secretory pathway proteins we identified Grasp55 in Cx36/Cx36-EGFP transfected conditions. B Streptavidin-affinity-captured proteins were detected on western blots using Streptavidin-HRP, anti-Cx36, anti-V5 and anti-Grasp55. C Immunoprecipitation using GFP trap demonstrates a PDZ dependent binding interaction between Cx36 and Grasp55-GFP. A Grasp55 variant lacking the PDZ binding motif fails to interact with Cx36. A Cx36 mutant lacking the PDZ binding motif does not co-precipitate with full length Grasp55-GFP. D Confocal scan of Cx36 and Grasp55 colocalizing in perinuclear structures. Line scan illustrates the overlap of intensity maxima. E Proximity ligation assay confirms a molecular association of Cx36 and Grasp55 that is diminished when the Cx36/S318ter mutant (lacking PDZ binding motif) is transfected. Difference in PLA labeling was highly significant. F Colocalization of PTPN13 and Cx36 at gap junctions in HEK293T cells. Scale: 10 µm
Grasp55 concentrates Cx36 in the GolgiTo further characterize the interaction between Cx36 and Grasp55, we first determined the subcellular compartment in which these proteins associate. In contrast to previous reports [34], we identified Grasp55 in the cis-Golgi as indicated by the colocalization with the Golgi matrix protein GM130. By contrast, the trans Golgi marker golgin97 showed no signs of association with Grasp55, suggesting that the scaffold protein is mainly restricted to the cis Golgi (Fig. 5A). Additionally, we observed some association of Sec24B and Grasp55, which is consistent with previous reports [20]. Co-expression of Cx36 and Cx36-EGFP combined with double labeling of organelle markers and Grasp55 further confirmed that Cx36 and Grasp55 colocalize in the cis Golgi, colocalized with GM130 (Fig. 5B). In the corresponding intensity scans a clear overlap for the maxima of all three channels can be seen. To investigate how Grasp55 impacts the vesicular transport of Cx36, we expressed the connexin in Grasp55 deficient HEK293T cells previously generated by Liu et al., [20] and quantified gap junction size, frequency and Cx36-containing ER whorls as an indicator of ER retention (Fig. 5C). Surprisingly, we found that Grasp55 deletion influenced none of these parameters. Compared to Cx36 transfected control HEK293T cells, Grasp55 KO cells showed no changes in gap junction size, frequency or ER whorls, indicating that Grasp55 is dispensable for the functional ER export of Cx36 and gap junction formation. While deletion of Grasp55 caused no apparent transport deficits, overexpression of a Grasp55-EGFP construct led to retention of Cx36 in perinuclear structures resembling the Golgi apparatus. This retention effect was prevented when the Grasp55 G2A mutant was transfected (Fig. 5D). The G2A mutant unlike wild type Grasp55 is not myristoylated at the N-terminus and fails to localize to the Golgi apparatus [35], suggesting that the retention effect of Grasp55 is caused by an interaction with the Golgi membrane. To test if Grasp55 affects the transition of Cx36 into the cis-Golgi we compared Cx36/Stx5 PLA reactivity between wild type and Grasp55 knock out cells. We observed no significant reduction in Cx36/Stx5 reactivity indicating that the Grasp55 deficiency does not affect the transition of Cx36 into the Golgi apparatus (Fig. 5E). Thus, we conclude that Grasp55 is dispensable for the ER export of Cx36 but could be necessary to concentrate the connexin in the Golgi apparatus.
Fig. 5
Grasp55 localizes to the cis Golgi where it can retain Cx36. A Grasp55 resides in the cis-Golgi and colocalizes with GM130. No colocalization can be detected in with trans-Golgi marker golgin97 or ER exit site marker Sec24B. B Grasp55 and Cx36, visualized as Cx36-EGFP/Cx36 heteromers. Detection of Cx36-EGFP via GFP allows immunolabelling of three channels, since mouse and rabbit antibodies were already used for organelle markers. Coexpression of native Cx36 is necessary to maintain functional PDZ interactions. C Knockout of Grasp55 does not compromise gap junction formation and Cx36 trafficking. Differences between conditions were not significant, P > 0.05. Significance was determined using a Mann–Whitney U test. D Overexpression of Grasp55 leads to retention of Cx36 in perinuclear structures. E PLA labeling of Stx5 and Cx36. Knockout of Grasp55 does not affect the transition of Cx36 into the Golgi. Difference in Cx36/Stx5 PLA intensity was not significant between wild type HEK294T cells and Grasp55 KO cells. P > 0.05. Significance was determined using a Mann–Whitney U test. Scale: 10 µm
留言 (0)