
記住我
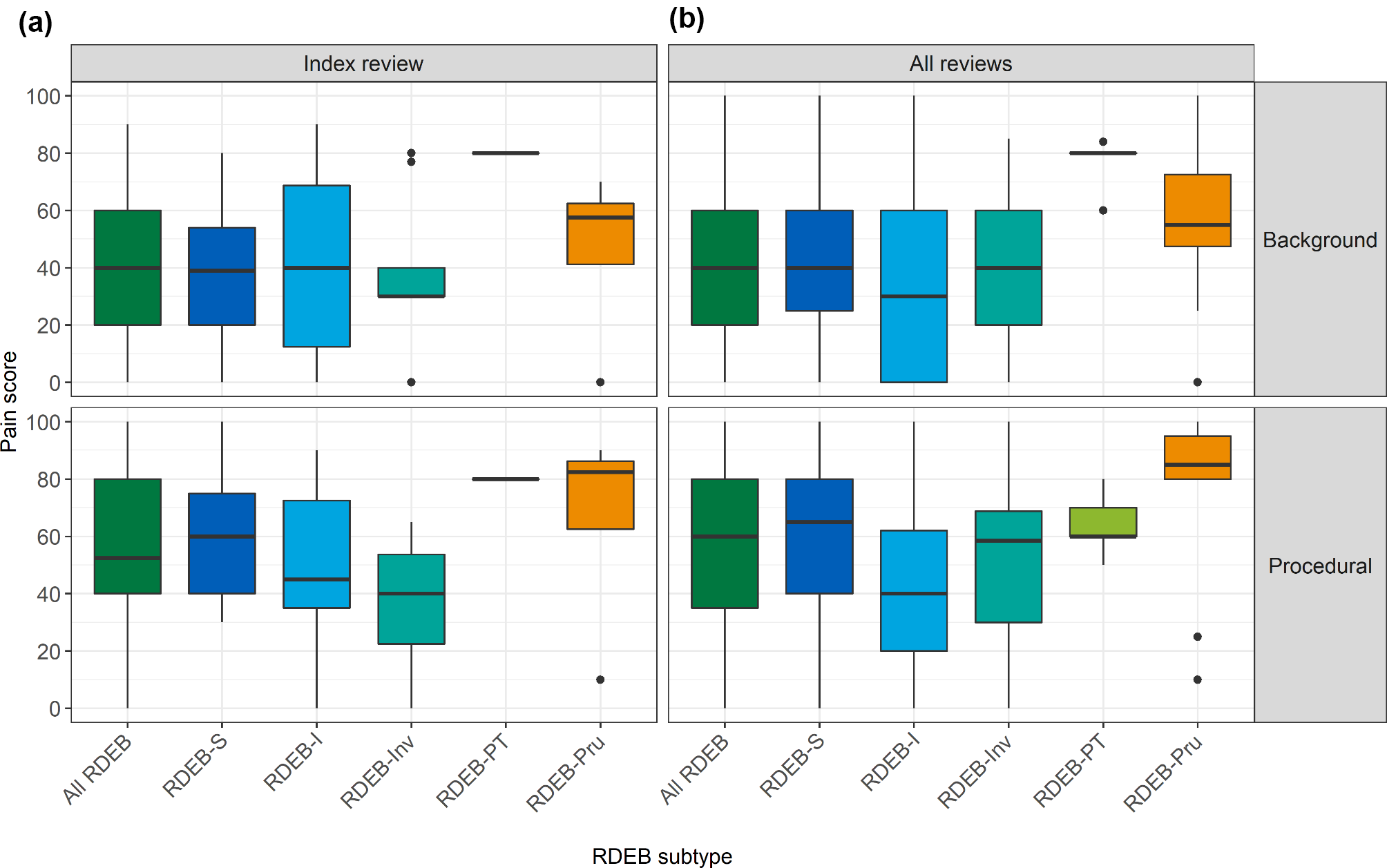






Patient 1 was an 8-month-old boy who was born to a healthy gravida 3, para 2 mother with a negative family history of genetic diseases. He was the first baby of this family, and his delivery at 40 weeks of gestation was uneventful except for obviously reduced fetal movement. He had normal birth measurements: weight 2.8 kg, length 50 cm and head circumference 33 cm. At 3.5 months of age, he exhibited axial hypotonia and could not raise his head. He displayed frequent blinking and nystagmus. Serum lactate level (2.69 mmol/L, normal range 0.5–1.7 mmol/L) was elevated. At 5 months of age, he suffered from intractable epileptic episodes characterized by staring, muscle rigidity, rapid blinking and autonomic symptoms. Electroencephalography (EEG) revealed rhythm weakness on the right side, with diffuse slow waves and epileptic discharges, particularly posteriorly. At 7 months of age, the patient had normal growth development: his height was 69 cm, and his weight was 9 kg. However, he had obvious microcephaly, and his head circumference was 39.5 cm (<-3 SD). Limb hypertonia manifested. Echocardiography revealed an atrial septal defect, diffuse hyperthermic cardiomyopathy and pericardial effusion. MRI showed bilateral underdeveloped insula, frontal, temporal and occipital lobes, microgyria, decreased white-matter volume, dilated bilateral lateral ventricles, bilateral symmetrical T2-weighted imaging (T2WI) multiple hyperintense lesions in the basal ganglia, thalamus and lenticular nucleus and a thin corpus callosum (Fig. 1A). His serum lactate level was increased (8.79 mmol/L). The patient passed away at 8 months of age due to respiratory failure.
Fig. 1
Brain MRI for our patients. Patient 1 (1 A): MRI showed bilateral underdeveloped insula, frontal, temporal and occipital lobes, microgyria, decreased white-matter volume, dilated bilateral lateral ventricles, bilateral symmetrical T2-weighted imaging (T2WI) multiple hyperintense lesions in the basal ganglia, thalamus and lenticular nucleus (a) and a thin corpus callosum (b); Patient 2 (1B): MRI showed frontal subdural fluid, bilateral hemispheric brain atrophy, multiple hyperintense lesions in the basal ganglia, globus pallidus, thalamus, midbrain and cerebral peduncles (a), and a thin corpus callosum (b); Patient 4 (1 C): Brain MRI revealed hyperintensity of the bilateral basal ganglia on symmetrical T2-weighted imaging (T2WI) (a) and a thin corpus callosum (b)
WES identified compound heterozygous variants, c.470 C > G, p.Thr157Arg and c.988dup, p.Arg330Lysfs*4, in TARS2 in the proband. The maternally inherited variant (c.470 C > G, p.Thr157Arg) has been previously reported in the literatures [10, 11]. The paternally inherited variant (c.988dup, p.Arg330Lysfs*4) is novel, and is predicted to truncate the aminoacylation domain of TARS2 (Fig. 2a). Both variants are classified as clinically pathogenic according to the ACMG/AMP guidelines [14].
Fig. 2
Variant identification by Sanger sequencing. Compound heterozygous TARS2 variants, c.470 C > G (p.Thr157Arg) (maternally inherited) and c.988dup (p.Arg330Lysfs*4) (paternally inherited), in patient 1 (a); c.470 C > G (p.Thr157Arg) (maternally inherited) and c.512G > A (p.Arg171Lys) (paternally inherited), in patient 2 and his sibling (patient 3) (b); a homozygous variant, c.470 C > G (p.Thr157Arg) in TARS2, in patient 4. Both parents were asymptomatic heterozygous carriers (c)
Family 2Patient 2 was the second child of a nonconsanguineous Chinese couple. The pregnancy was normal except for significantly reduced fetal movement. He was born at full term with normal birth measurements: his length was 49 cm, his weight was 3.64 kg and his head circumference was 34 cm. His Apgar scores were 8, 9, and 9. After birth, he was referred to the clinic due to pneumonia and jaundice. At 1 month of age, he displayed axial hypotonia and feeding difficulties. At 2.5 months of age, he was diagnosed with focal epilepsy, with each seizure lasting for approximately one minute and characterized by staring and rigidity of the upper extremities. EEG showed epileptiform discharges with frequent polyspikes and slow waves. The symptoms could not be effectively alleviated by administering antiepileptic drugs. Serum lactate (7.03 mmol/L) and blood NH3 (73 µmol/L, normal range 9–47 µmol/L) levels were elevated. At 4 months of age, he developed limb hypertonia. MRI showed frontal subdural fluid, bilateral hemispheric brain atrophy, multiple hyperintense lesions in the basal ganglia, globus pallidus, thalamus, midbrain and cerebral peduncles, and a thin corpus callosum (Fig. 1B). He had a growth delay at 6 months of age: his height was 63 cm (<-2 SD), his weight was 6.2 kg (<-2 SD), and his head circumference was 40 cm (-3 SD). He died due to severe respiratory failure at 1 year and 10 months of age.
His elder brother (Patient 3) was also born after an uneventful pregnancy, during with markedly reduced fetal movement was observed. His birth measurement parameters were normal: his weight was 2.6 kg, length was 50 cm, and head circumference was 34 cm. He also exhibited clinical features of mitochondrial encephalopathy that were strikingly similar to those of his younger brother. He had never achieved the ability to raise his head alone and exhibited no eye contact. Increased serum lactate level was regularly monitored. He exhibited delayed growth: his height was 73 cm (<-2 SD), his weight was 9 kg (<-2 SD), and his head circumference was 42.5 cm (<-3 SD) at 1 year and 3 months of age. He died due to respiratory problems, lactic acidosis and severe developmental delay at 2 years and 4 months of age.
WES revealed the recurrent missense variant, c.470 C > G, p.Thr157Arg (maternally inherited), and a novel variant, c.512G > A, p.Arg171Lys (paternally inherited), in TARS2 in the two affected children. The variant c.512G > A, p.Arg171Lys was located in the N2 domain and was highly conserved among different species, and was absent in in the Genome Aggregation Database (Fig. 2b). The variant was confirmed in trans with the previously identified pathogenic variant c.470 C > G, p.Thr157Arg. Thus, the variant was classified as likely pathogenic according to the ACMG/AMP guidelines [14].
Family 3Patient 4 was a 2-year and 2-month-old female born to unrelated parents. She was born at full term after an uneventful pregnancy. She had normal birth measurements: her length was 50 cm, her weight was 3.3 kg, and her head circumference was 33 cm. She was able to raise her head at 5 months of age. However, the patient presented with progressive developmental regression, including unstable head control and axial hypotonia at 7 months of age, and she gradually displayed limb spasticity after eight months of age. She had obvious feeding difficulties. An increased serum lactate level was detected (4.9 mmol/L). Furthermore, plasma amino acid analysis revealed increased free carnitine (70.28 µmol/L, normal range 9-53.7 µmol/L) and hyperammonemia (98 µmol/L, normal range 18–72 µmol/L). At 5 months of age, she displayed infantile spasms, characterized by brief generalized muscle spasm with straight arms extended and bent torso and legs. The events lasted for 15 s–1 min each time, and eight episodes usually occurred every day. EEG revealed hypsarrhythmia, including a large number of irregular high-voltage, sharp spikes/polyspikes/slow waves. The patient’s epilepsy was not controlled even when sodium valproate or a ketogenic diet was applied. The serum lactate concentration was maintained at a constant level according to real-time monitoring. Brain MRI revealed hyperintensity of the bilateral basal ganglia on symmetrical T2-weighted imaging (T2WI) and a thin corpus callosum at 7 months of age (Fig. 1C). On recent physical examination at 2 years and 2 months of age, she exhibited growth delay: her height was 82 cm (-2 SD), her weight was 10 kg (-2 SD), and her head circumference was 43.5 cm (<-3 SD). She was not able to raise her head and had no language or cognitive development.
WES revealed a homozygous variant, c.470 C > G, p.Thr157Arg in TARS2, in the patient. Both parents were asymptomatic heterozygous carriers (Fig. 2c).
留言 (0)