
記住我


A 15-year-old young boy was admitted to our department due to proteinuria and hematuria with elevated blood pressure (170/120 mmHg) for 1 week. The laboratory examination showed macrocytic anemia (hemoglobin 9.3 g/dl, MCV 105.3 fl.), proteinuria (3+), and hematuria (20–30 cells/HPF). The 24-hour urinary protein was 2.12 g/day (total urine volume 500 ml) with decreased serum albumin level of 34.4 g/L. Serum creatinine (SCr) was 110.2 µmol/L (reference range: 56–102 µmol/L). Serum level of homocysteine (HCY) was over 200 µmol/L (reference range: 6–17 µmol/L), which exceeded the detection limit in routine test. He had a comparatively low level of complement component (C3 0.587 g/L, reference range: 0.6–1.5 g/L; C4 0.106 g/L, reference range: 0.12–0.36 g/L) and a low titer of positive antinuclear antibody (ANA) (1:100 ). However, no systemic immune or infection related disease was observed. He had impaired urine concentration function, with significantly low morning urinary osmotic pressure (310 mOsm/kg, reference range: 600–1000 mOsm/kg), and blood osmotic pressure was normal (293 mOsm/kg, reference range: 275–305 mOsm/kg). Urine glucose was negative and urine acidification examination was normal, without hypophosphatemia and hypouricemia. Schistocytes on the blood smear could be seen, accounting for 1.2%, with thrombocytopenia (platelet 118,000/mm3), and increased lactate dehydrogenase (LDH 573 IU/L), which indicated the microangiopathic hemolytic anemia (MAHA), without any history of thrombosis. However, von Willebrand factor protease (ADAMTS13) and the concentration of factor H were both normal, and the anti-factor H antibody was negative..
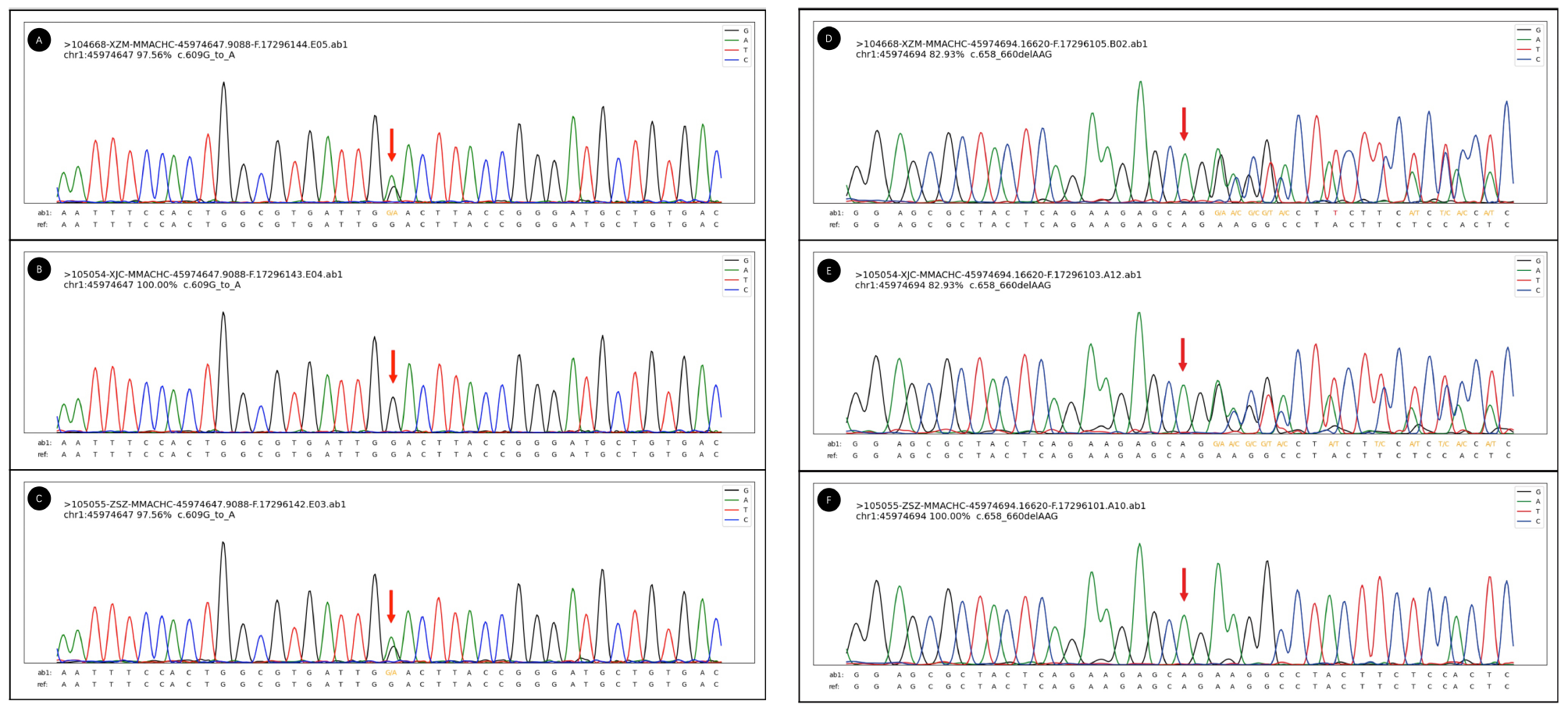
He was diagnosed with cblC 2 months after his birth, and treated with hydroxocobalamin, L-carnitine and betaine ever since. Comprehensive genetic analyses revealed compound heterozygous variants c.609 G > A (p.W203*) (The American College of Medical Genetics and Genomics, ACMG classification: pathogenic) and c.658_660del (p.K220del) (ACMG classification: likely pathogenic) mutation in the MMACHC gene (Fig. 1). He developed well in spite with stable disease. He had no signs of involvement of intelligence, sensibility or central nervous system symptoms, such as seizures, ataxia, or hypotonia, until he interrupted his therapy 4 months before admission.
Fig. 1
Sanger electropherogram figure of MMACHC mutation in patient 1. A to C and D to F shows the mutated locus of patient 1, his father, and his mother on c.609 G > A (p.W203*) and c.658_660del (p.K220del), separately
A renal biopsy was performed (Fig. 2). Light microscopic examination showed that 1/28 glomeruli was ischemic and sclerotic. The rest of the glomeruli showed mild mesangial expansion, accompanied by segmental endothelial cell proliferation. The glomerular basement membrane thickened diffusely, with the formation of “double contours”. The tubules displayed loss of brush border with epithelial simplification. Direct immunofluorescence revealed trace staining for segmental granular deposition of IgA (+) and IgM (+) on mesangial area, and negative for IgG, C3, and C1q. He was diagnosed with renal TMA and acute tubular injury as the renal complication of cblC. Electron microscopy confirmed the diagnosis, and no enlarged mitochondria was detected. The patient was treated with hydroxocobalamin (3 mg/day, i.m.), betaine (9 g/day, p.o.), L-carnitine (2 g/day, p.o.) as well as Vitamin AD and vitamin B compounds daily. Besides, angiotensin receptor blockers and calcium channel blockers combinations were used to treat hypertension. After 6 weeks of treatment, his blood pressure, hemoglobin, LDH and urine protein returned to normal. HCY decreased to 85.56µmol/L and SCr decreased to 65 µmol/L which indicated that an event of acute kidney disease occurred in this patient.
Fig. 2
Kidney biopsy findings. (A) The glomerulus exhibited segmental endothelial cell proliferation with diffusely thickened glomerular basement membrane and the formation of ‘double contours’, and a granuloma consisted of mainly foam cells at beside interstitium (PAS ×400). (B) The loss of brush border with epithelial simplification of tubular epithelia was presented, along with focal infiltration of lymphocyte and mononuclear cells in renal interstitium (HE×400). (C) Electron microscopy showed widening of subendothelial area of glomerular basement membrane with narrowed cavity of capillary loops (original magnification ×5,000). (D) Proliferation of mesangial cells and endothelial cells with occlusion of glomerular capillary lumen was observed (original magnification×6,000)
Patient 2A 15-year-old boy was admitted to our department due to proteinuria and hematuria for 1 year. His blood pressure was 135/85 mmHg. Urinary protein excretion was 2.05 g/day (urine volume 1000 ml) and his SCr was 72 µmol/L (reference range 44–133 µmol/L). Urinary N-acetyl-beta-D-glucoaminidase (NAG) was elevated to 18.7 U/L. HCY was 149.01 µmol/L, which was also significantly increased. Urine glucose was negative, along with hyperuricemia but normal serum phosphorus. ANA was positive (1:320). Anti-dsDNA, anticardiolipin antibodies, and anti-β2-GP1 antibodies were all negative. Echocardiography showed an enlarged right heart with pulmonary arterial hypertension (86 mmHg). The level of hemoglobin and platelet count were both normal. There were no evidence of MAHA and no signs of thrombotic event in examinations. Renal biopsy showed TMA, combined with focal segmental glomerulosclerosis (FSGS) like lesions. Tubular epithelial cells presented with vacuolar and granular degeneration.
He went to the other medical center due to dyspnea 1 year ago. During hospitalization, examination showed pulmonary arterial hypertension and megaloblastic anemia. Electromyography (EMG) and nerve conduction velocity (NCV) showed peripheral nerve injury in the upper and lower limbs. Genetic analyses showed compound heterozygous variants of the c.80 A > G (p.Q27R) (ACMG classification: pathogenic) and c.609G > A (p.W203X) (ACMG classification: pathogenic) in MMACHC gene, which confirmed the diagnosis was MMA-uria and hyperhomocysteinaemia (cblC type). The patient was 195 cm in height and weighed 70 kg but his physical strength was worse than his peers, and he had not attended physical education classes since the fifth grade of primary school. Intelligence Quotient (IQ) test showed he has a normal IQ score of over 90.
Treatment with mecobalamin (3 mg/day, p.o.), cobamamide (3 mg/day, i.m.), folic acid (5 mg/day, p.o.), betaine (9 g/day, p.o.), L-carnitine (2 g/day, p.o.), as well as Vitamin AD compounds and vitamin B supplements. Lotensin (10 mg/day, p.o.) was used to anti-hypertension and ambrisentan (5 mg/day, p.o.) was also used to reduce pulmonary artery pressure. Within the next 10 days, the serum HCY was decreased to 136.22 µmol/L and 24 h-urinary protein excretion was reduced to 1.18 g/day (total urine volume 650 ml). At the last follow-up, two months after the discharge, the patient’s serum HCY was decreased to a satisfactory level of 50.13 µmol/L, and blood pressure was well controlled under antihypertensive agents.
Patient 3A 16-year-old boy without significant medical history presented with severe hypertension (up to 180/116 mmHg) with the chief complaint of a decrease in activity tolerance, mucosal hemorrhage, and oliguria. Further examination showed anemia (Hb 6.3 g/dl) with schistocytes, thrombocytopenia (90,000/mm3), kidney injury (SCr 197 µmol/L), proteinuria, elevated lactate dehydrogenase (LDH 779 U/L) and abnormal NT-proBNP (6152 pg/ml). Ultrasonography revealed enlargement of left atrial and the thickening of left ventricular wall and splenomegaly. Renal biopsy showed typical signs of TMA in glomerulus, which presented as duplication of the glomerular basement membrane (GBM), endothelial swelling, mesangiolysis and intraglomerular thrombi, along with swelling of the tubular epithelium, and a patchy infiltration of lymphocytes and monocytes was present within the interstitial area, with interstitial fibrosis. ANA, serum complement level, H factor and ADAMTS 13 activity were normal. There was no evidence of any bacterial and viral infection, including HIV, as well as the effect of drugs or toxic exposure. Glucocorticoid, plasmapheresis and antihypertensive drugs (including RAASi) were used for TMA therapy. However, although hemolysis was rapidly controlled with normalization of the LDH and platelet count, his kidney injury was still progressive to end-stage kidney disease. In addition, he also presented with renal tubular acidosis and persistent hyperkalemia. This phenomenon forced us to continue searching for the cause of TMA.
The intellectually disabled sister in family history and a marked rise in serum level of HCY (247.8 µmol/L) put the focus on cobalamin C metabolism. A higher level of urine HCY (65.71 µmol/L) supported our suspicion. Nutritional B12 deficiency was excluded by a normal serum level. However, chromatographic evaluation of the urine showed methylmalonic aciduria. A cobalamin C (cblC) deficiency presented with MMA-uria and hyperhomocysteinaemia was subsequently confirmed by genetic testing of the two heterozygous likely pathogenic mutations of c.1 A > G (p.Met1?) (ACMG classification: likely pathogenic) and c.80 A > G (p.Q27R) (ACMG classification: likely pathogenic) in the MMACHC gene. The same genetic mutation was also found in the blood sample of his mother and father, respectively.
After a definite diagnosis of cblC deficiency, the patient was treated with hydroxocobalamin, betaine, calcium folinate and L-carnitine. After one month of therapy, serum HCY decreased to 83 µmol/l, and urine HCY decreased to 28 µmol/l, while the patient was still hemodialysis-dependent. It was reassuring that after 10 months of drug therapy, the level of hemoglobin, platelet and LDH recovered. Furthermore, this patient was finally dialysis-free with the level of SCr maintained at185 µmol/L.
留言 (0)