
記住我






Overall, 159 participants (n = 133, immediate rollover; n = 26, delayed rollover) received ≥ 1 dose of viloxazine ER and were included in safety and efficacy assessments (Table 1). The mean ± standard deviation (SD) exposure to viloxazine ER in the open-label extension was 265 ± 254.9 days (median 191 days; range 1–887 days). Notably, 81 participants (50.9%) received treatment for at least 6 months, 50 (31.4%) received treatment at least 12 months, and 9 (5.7%) received treatment for longer than 24 months (Fig. 1). Mean ± SD participant age was 36.3 ± 10.3 years, with enrollment balanced between male (51.6%) and female (48.4%; Table 2) participants. Overall, 108 (67.9%) participants reported taking concomitant medications during the study. The most common (> 5%) medications were vitamins; contraceptives; viral vaccines; and single doses or short courses of analgesics (e.g., ibuprofen, acetaminophen), antihistamines (e.g., cetirizine, loratadine, diphenhydramine), or antibiotics. Additionally, ten participants (6.3%) were prescribed a stimulant medication (methylphenidate or amphetamine product) concomitantly at some point during the trial. Nearly all participants (90%) reported caffeine consumption while taking viloxazine ER, with 29.6% consuming ≥ 1000 mg of caffeine weekly.
Table 1. Participant dispositionFig. 1
Exposure duration to viloxazine ER.
Table 2. Demographics and baseline characteristicsParticipants were started at 200 mg/day viloxazine ER; after week 1 (start of week 2), 103 participants received at least 400 mg/day of viloxazine ER. Following initial dose optimization (≥ week 13), the most frequently used dose was between 400 and 600 mg/day for most participants (73%), with 36% using 600 mg/day (Fig. 2, Supplementary Fig. 2).
Fig. 2
Modal dose used following optimization (≥ week 13).
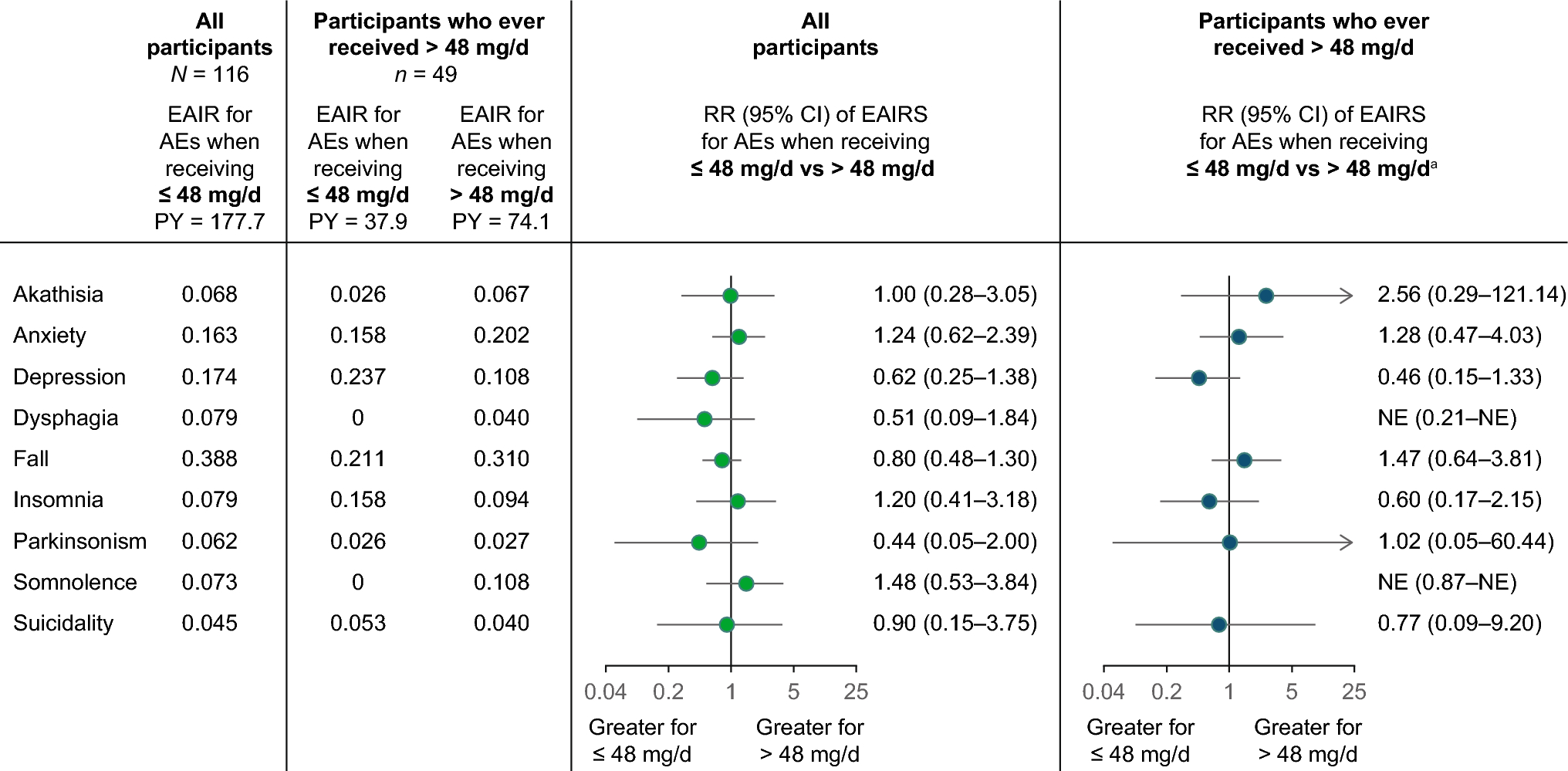
3.2 Primary Outcome: Safety3.2.1 Adverse EventsOverall, 72.3% (n = 115) of participants experienced 1 or more AEs during the open-label extension, most commonly insomnia (13.8%), nausea (13.8%), headache (10.7%), and fatigue (10.1%) (Table 3). All of these were reported more frequently for participants who had previously received placebo in the prior double-blind trial (Table 3). For 51.6% (n = 82) of participants, ≥ 1 AEs were considered to be related to viloxazine ER treatment, most commonly (≥ 5% occurrence) insomnia (11.3%), fatigue (10.1%), nausea (9.4%), headache (5.7%), and dry mouth (5.0%). For most participants, AEs were mild (26.4%) or moderate (40.3%) in severity. Of the 5.7% reporting any severe AE, the only events reported by more than 1 individual were insomnia (n = 2) and headache (n = 2).
Table 3. Summary of adverse eventsTwo participants (1.3%) experienced serious AEs (SAEs), all of which were deemed by investigators to be severe, but unrelated to study medication. One of these participants experienced serious adverse events of deep vein thrombosis and pulmonary embolism on study day 97 that resolved but resulted in treatment discontinuation. The second participant experienced serious adverse events of fall, spinal column injury, and syncope on study day 341 but remained in the study without a change in viloxazine ER dose. Syncope was reported as an AESI for two participants (1.3%). These included the participant with the SAE (described above), and a participant who experienced moderate syncope on study day 146 that was also assessed as unrelated to viloxazine ER and did not require a change in dosage.
Overall, AEs led to treatment discontinuation for 28 (17.6%) total participants, including 20 (22.5%) of the 89 who had received placebo during the double-blind trial and 8 (11.4%) of the 70 who had received viloxazine ER during the double-blind trial. The most common AEs leading to treatment discontinuation were insomnia (2.5%), nausea (2.5%), and fatigue (1.9%). Of the ten participants who received supplemental stimulant treatment, three (33.3%) reported onset of any AE during stimulant use (participant 1: anxiety and sinusitis; participant 2: nausea and irritable bowel syndrome; and participant 3: somnolence and an episode of food poisoning). None of these AEs were severe or serious or resulted in viloxazine ER discontinuation.
3.2.2 Clinical Laboratory Evaluations, Vital Signs, Physical Examinations, Electrocardiograms, and SuicidalityClinical hematology laboratory monitoring showed no new safety signals. A total of four (2.5%) participants had elevations in aspartate aminotransferase (AST; n = 3) and/or alanine aminotransferase (ALT; n = 4) that were reported as AEs. Viloxazine ER was withdrawn for two of these participants (both of whom had received viloxazine ER during double-blind without clinically significant transaminase elevations and were participating in the open-label under the delayed-entry protocol). Elevations were < 3× the upper limits of the reference ranges (female/male: AST 34/39 U/L and ALT 33/44 U/L, respectively) in all but one individual, a 37-year-old man with AST 54 U/L and ALT 151 U/L on study day 91 (considered possibly related to treatment) that led to viloxazine ER discontinuation. The other participant who discontinued was a 55-year-old woman who had ALT and occasional AST elevations prior to both double-blind and open-label treatment; AST/ALT remained elevated during treatment with the highest reported values of 43/84 on day 23 that were reported as an AE of moderate elevated liver enzymes (considered unlikely related to treatment), and the individual was subsequently withdrawn from the study due to this AE. All AST/ALT elevations resolved, including for the 2 participants who continued study medication without interruption, and none were associated with increases in bilirubin or alkaline phosphatase.
Elevations in systolic blood pressure, diastolic blood pressure, and heart rate were also observed during the study, with mean ± SD elevations (measured while sitting) at participants’ last on-study visit (LOSV) of 2.5 ± 11.2 mmHg, 1.5 ± 8.3 mmHg, and 4.8 ± 11.9 beats/min, respectively. Of the 153 participants for whom measurements were available, 30.7% (n = 47) and 14.4% (n = 22) had any sitting systolic or diastolic blood pressure increase of ≥ 15 mmHg, and 24.8% (n = 38) had any post-baseline heart rate increase of ≥ 20 beats/min at any time during the open-label extension trial. However, these increases may not have been sufficiently high nor persistent to meet a definition of hypertension. In fact, blood pressure or heart rate increases were not commonly reported as AEs, and only seven participants (4.4%) reported any AE related to elevated blood pressure [hypertension (n = 3), increased diastolic blood pressure (n = 3), or increased blood pressure (n = 1)], and five total participants (3.1%) reported AEs of increased heart rate and/or tachycardia (including two participants with tachycardia on ECG at a single visit where the AE was considered mild and unlikely/not related to treatment). All AEs of increased blood pressure or heart rate were mild or moderate in severity, and none resulted in dose modifications or treatment discontinuation. Additionally, three participants discontinued for AEs considered cardiac in nature, one for palpitations (moderate, treatment-related), one for irregular heart rate (mild; possibly treatment-related), and one for sinus arrhythmia (moderate, unlikely treatment-related) that was noted on ECG. The specific arrhythmia was not reported for this individual; however, all ECG intervals were within normal ranges and no treatment was administered for the event.
A total of three (1.9%) participants reported bodyweight changes as an AE [weight increased (n = 1), unlikely treatment-related; weight decreased (n = 2), possibly treatment-related] that were mild in severity and did not result in treatment change or discontinuation. Overall, the mean ± SD change from baseline in body weight at participants’ LOSV was – 0.5 ± 4.95 kg. Of 153 participants with available data, 14.4% (n = 22) experienced weight gain of ≥ 5% of their bodyweight, and 27.5% (n = 42) experienced a loss of ≥ 5% at any study visit. At participants’ LOSV, these percentages were 9.8% (n = 15) and 17.0% (n = 26), respectively.
Two participants (1.3%) reported suicidal ideation on C-SSRS, with positive responses to the question, “Have you wished you were dead or wished you could go to sleep and not wake up?” For the first participant, the response was recorded on study day 14 and was not reported as an AE; for the second participant, the response was recorded on study day 644 and was reported as an AE that was assessed as mild and not treatment-related. Neither participant discontinued or had a viloxazine ER dose change as a result of the response, and both continued participation through the end of the study (days 704 and 867, respectively). A third participant recorded a positive response to this same C-SSRS question prior to beginning open-label viloxazine ER treatment (visit 1); this participant subsequently quit taking viloxazine ER on day 8 due to AEs of insomnia, dry mouth, and restless leg syndrome. No suicidal ideation was reported at the early termination visit. There were no other “yes” responses to C-SSRS questions. No participant reported suicidal behavior (neither on C-SSRS nor as an AE).
3.3 Secondary Outcome: EfficacyOverall, ADHD symptoms showed consistent improvement over the course of the trial, beginning at the first clinic assessment (week 2), where the mean ± SD change from baseline (37.9 ± 6.3) in AISRS total score was –11.4 ± 9.5 points (Fig. 3). AISRS scores by this timepoint were similar regardless of prior treatment group or rollover status, with absolute scores of 26.1 ± 10.5 (n = 95) for delayed rollover participants and those treated with placebo in the double-blind trial versus 27.4 ± 12.8 (n = 51) for immediate rollover participants treated with viloxazine ER in double-blind (Fig. 3). Further improvement was seen at subsequent visits, with overall AISRS score reductions at 6 months (week 28), 1 year (week 52), and LOSV of ‒ 19.9 ± 9.9 (n = 81), ‒22.0 ± 10.4 (n = 51), and –18.2 ± 11.5 (n = 146), respectively. Improvement was observed for both AISRS Inattention and Hyperactivity/Impulsivity subscales, which also decreased from baseline scores of 21.6 ± 3.4 and 16.2 ± 4.9, respectively, by week 2 (‒ 6.1 ± 5.6 and – 5.3 ± 5.4), with further reductions (improvement) at subsequent visits (Supplementary Table 1). Improvement in these subscales at 6 months (week 28), 1 year (week 52), and LOSV were, respectively,– 11.2 ± 5.9, – 11.8 ± 6.0, and – 9.9 ± 7.1 for Inattention, and – 8.7 ± 5.3, – 10.2 ± 5.8, and – 8.3 ± 5.9 for Hyperactivity/Impulsivity.
Fig. 3
AISRS total score change from baseline by visit. For participants who enrolled in the open-label trial within 7 days of completing the previous double-blind trial (immediate rollover), BL was the double-blind BL score; for participants who enrolled in the open-label trial more than 7 days after the previous double-blind clinical trial (delayed rollover), BL was measured immediately prior to the first dose of viloxazine ER in the open-label study. The inset shows the improvements over the first 20 weeks of the open-label extension for immediate rollover participants who received viloxazine ER in the double-blind trial and delayed rollover participants who received viloxazine ER during the double-blind trial or immediate/delayed rollover participants who received placebo. ADHD, attention-deficit/hyperactivity disorder; AISRS, ADHD Investigator Symptom Rating Scale; BL, baseline; CFB, change from baseline; DB, double-blind; ER, extended-release; LOSV, last on-study visit; SD, standard deviation
At the end of the double-blind trial, 60.0% (n = 78/130) and 39.2% (n = 51/130) of participants receiving viloxazine ER had met the AISRS 30% and 50% reduction responder thresholds, respectively. Following dose optimization in the open-label trial (week 12), the percentage of participants meeting these responder thresholds at all subsequent visits was above 70% for the AISRS 30% reduction requirement, and above 50% for the AISRS 50% reduction requirement. The generally high responder rates were not solely the result of dropout among nonresponders, in that 71.9% (n = 105/146) of participants who had any post-baseline data experienced a ≥ 30% reduction in AISRS total score at their LOSV, and that 47.3% (n = 69/146) of participants (Fig. 4) experienced a ≥ 50% reduction at their LOSV.
Fig. 4
AISRS total score ≥ 50% responder rates by visit. For participants who enrolled in the open-label trial within 7 days of completing the previous double-blind trial (immediate rollover), BL was the double-blind BL score; for participants who enrolled in the open-label trial more than 7 days after the previous double-blind clinical trial (delayed rollover), BL was measured immediately prior to the first dose of viloxazine ER in the open-label study. AISRS 50% responder rate is defined as the percent of participants at each visit who had a ≥ 50% reduction from BL in AISRS total score. ADHD, attention-deficit/hyperactivity disorder; AISRS, ADHD Investigator Symptom Rating Scale; BL, baseline; DB, double-blind; ER, extended-release; LOSV, last on-study visit
CGI-S and CGI-I results mirrored the improvement in AISRS scores. The CGI-S score was 4.6 ± 0.6 at baseline (all participants at least moderately ill), improved by – 1.0 ± 1.0 at week 2, and showed further reductions at subsequent visits. Reductions from baseline at 6 months (week 28), 1 year (week 52), and LOSV were –1.8 ± 1.2, –2.1 ± 1.3, and –1.6 ± 1.3, respectively (Fig. 5). At LOSV (n = 159), 56.6% of participants had improved such that they would be classified as between not at all ill to mildly ill, while 43.4% remained moderately to severely ill. CGI-I ratings also reflected improvement at week 2 (score 2.9 ± 0.96) and decreased (improved) further at subsequent study weeks. Scores at 6 months (week 28), 1 year (week 52), and LOSV were 2.2 ± 0.9, 1.9 ± 1.1, and 2.4 ± 1.1, respectively (Table 4, Supplementary Fig. 3A). At LOSV (n = 152), 23.0% of participants were very much improved, 28.3% were much improved, 27.6% were minimally improved, 18.4% had no change, 2.0% were minimally worse, and 0.7% were much worse.
Fig. 5
CGI-S total score by visit. For participants who enrolled in the open-label trial within 7 days of completing the previous double-blind trial (immediate rollover), BL was the double-blind BL score; for participants who enrolled in the open-label trial more than 7 days after the previous double-blind clinical trial (delayed rollover), BL was measured immediately prior to the first dose of viloxazine ER in the open-label study. The inset shows the improvements over the first 20 weeks of the open-label extension for immediate rollover participants who received viloxazine ER in the DB trial and delayed rollover participants who received viloxazine ER during the double-blind trial or immediate/delayed rollover participants who received placebo. BL, baseline; CGI-S, Clinical Global Impression-Severity of Illness; DB, double-blind; ER, extended-release; LOSV, last on-study visit; SD, standard deviation
Table 4. Summary of the absolute values at baseline and by visit for CGI-I and AAQoL measures and change from baseline in AISRS and BRIEF-A scales by visitAs with AISRS scores, participants who had received placebo in double-blind and those who returned to the trial as delayed-rollover participants showed rapid improvement upon starting viloxazine ER treatment. By week 2, CGI-S scores for these participants were similar to direct rollover participants who had received viloxazine ER during double-blind treatment (3.6 ± 1.03 and 3.6 ± 1.03, respectively).
CGI-S responder rates and CGI-I responder rates on viloxazine ER treatment reached 30.8% (n = 40/130) and 48.5% (n = 63/130), respectively, at the end of the double-blind trial and continued to increase during open-label treatment. Respective CGI-S and CGI-I responder rates were 29.5% and 61.6% following initial dose optimization (week 12), 42.0% and 70.4% at 6 months (week 28), and 56.9% and 80.4% at 1 year (week 52), and generally remained at or higher than these levels at subsequent visits. At participants’ LOSV (n = 146), 37.0% were classified as CGI-S responders and 71.9% as CGI-I responders (Fig. 6, Supplementary Fig. 3B). A similar pattern of improvement was observed for other secondary efficacy endpoints throughout the open-label extension trial, including BRIEF-A GEC, BRI and MI index T-scores, AAQOL, GAD-7, and SDQ (Table 4, Supplementary Table 1).
Fig. 6
CGI-S responder rates by visit. For participants who enrolled in the open-label trial within 7 days of completing the previous double-blind trial (immediate rollover), BL was the double-blind BL score; for participants who enrolled in the open-label trial more than 7 days after the previous double-blind clinical trial (delayed rollover), BL was measured immediately prior to the first dose of viloxazine ER in the open-label study. CGI-S responder rate is defined as the percent of participants at each visit who had a CGI-S score of 1 (normal, not at all ill, asymptomatic) or 2 (borderline ill). BL, baseline; CGI-S, Clinical Global Impression-Severity of Illness; DB, double-blind; ER, extended-release; LOSV, last on-study visit
留言 (0)