
記住我
Staphylococcus aureus infections are not new to the world; they are responsible for high death rates due to hospital-acquired infections. Patients admitted to intensive care units, patients with implants, and patients in the postsurgical unit are among the major groups affected by virulent strains of Staphylococcus aureus or methicillin-resistant Staphylococcus aureus (MRSA) (Foster, 2004; Sampedro and Bubeck Wardenburg, 2017; Troeman et al., 2023). The prevalence of diseases caused by MRSA is not confined to hospitals; it is a major pathogen isolated from community-acquired infections (Berman et al., 1993; DeLeo et al., 2010). It is associated with suppurative infections, skin and soft tissue infection, pneumonia, endocarditis, food poisoning, surgical site infection, etc (Lowy, 1998; Foster, 2004; Tong et al., 2015).
The gradual increase in antibiotic resistance in Staphylococcus aureus is a point of concern to the existing healthcare system across the world. It contributes to the pathogenicity and virulence of this bacterium (Otto, 2010). It is considered the most common pathogen with drug resistance and a very high mortality rate following nosocomial infections (Jarvis et al., 2007; Klein et al., 2007; Klevens et al., 2008). The increasing rates of drug resistance development have an impact on the global burden of infections with high severity. This may lead to the exhaustion of antibiotic options for treating this pathogen. Currently, there is a demand for an alternative therapy for most pathogenic strains of S. aureus that are not susceptible to existing antibiotics due to drug resistance (Sampedro and Bubeck Wardenburg, 2017; Prencipe et al., 2022). In recent reports, due to the lack of many drug options on the market, newer drugs were under clinical trials; however, the cost for development and testing of only 59 drugs during 2015-2016 was estimated to approximately 19 million dollars, which was a high cost for conventional drugs which bacteria may turn resistant to in future (Moore et al., 2018).
The adhesin proteins present on the surface of Staphylococcus play a significant role in the adherence of the pathogen to host cells. This process is mediated through a receptor on the surface of host cells, also known as a ligand. These proteins belong to a family or group of proteins called MSCRAMM (microbial surface components recognizing adhesive matrix molecules) that represent the surface component of the bacterial cell. Surface adhesin proteins participate in colonization as well as infection (Patti et al., 1994; Peacock et al., 2002). Some important adhesin proteins of Staphylococcus aureus are clumping factor (Clf), biofilm-associated protein (Bap), collagen binding protein (Cna) and fibronectin binding protein (FnBP) (Walsh, 2000; Mulcahy et al., 2012; Soltani et al., 2019). These proteins are associated with several virulence factors and are responsible for the multifaceted virulence of S. aureus. The formation of biofilms, bacterial accumulation in plasma, cardiovascular diseases through platelet activation, attack on immune cells, etc., are some of the contributions of surface adhesins to staphylococcal infection (Paharik and Horswill, 2016; Josse et al., 2017; Soltani et al., 2019).
Due to increasing resistance to antibiotics and the challenges in treating S. aureus and MRSA, alternative methods to develop drugs need to be explored. In this study, we adopted the principle of structure-based drug discovery to develop an antagonist that interrupts ligand-adhesin binding. We selected the clumping factor adhesins ClfA and ClfB for this study because of their roles in both colonization and infection. By molecular docking, various candidates were selected based on their binding to the active sites of ClfA and ClfB. Later, the interactions and properties between the shortlisted compounds and ClfA and ClfB were analyzed by molecular dynamics. We also studied the drug likeness of the selected compounds.
2 Materials and methods2.1 Structure-based drug designAfter a thorough review of the mechanism and properties of the clumping factor components ClfA and ClfB, their 3D structures were obtained from a protein bank library (www.rcsb.org) (Hawkins et al., 2012). The structure was further refined and processed by the PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) and minimized using UCSF Chimera version 1.14 software (23). Next, a standard, allantodapsone, was identified for comparison of the identified compound, its interaction pattern with the protein and its drug likeness. The physiochemical properties of allantodapsone were studied in detail at https://pubchem.ncbi.nlm.nih.gov (Prencipe et al., 2022). Compounds similar to allantodapsone were identified from a bank of approved drugs and antimicrobial agents (Frog V 2.14). In our study, the Aurora FC Antibacterial database and DrugBank were utilized for identifying ligand compounds.
Drug likeliness and toxicity analyses of the selected compounds/ligands were performed using SwissADME (http://www.swissadme.ch/) (Sarkar, 2021), whereas toxicity analysis was performed based on parameters such as hepatotoxicity, carcinogenicity, mutagenicity and cytotoxicity using ProTox-II (http://tox.charite.de/protox_II) (Banerjee et al., 2018).
Molecular docking of the 3D structures of ClfA and ClfB with standard and shortlisted compounds was performed by using Autodock 4.2 (Morris et al., 2009). The active site composition of amino acids was determined in a previous study (Prencipe et al., 2022). During docking, the amino acids at the active site of the adhesin proteins (ClfA and ClfB) were kept rigid, while the ligand molecules were allowed to change positions. The PDBQT files of the proteins and ligands were prepared with AutoDock Tools (v.1.5.6) of the MGL software package. The protein-ligand docking complex was analyzed by the Lamarckian genetic algorithm (LGA) method.
The 2D and 3D structures of the docking complex were studied for interpreting the binding energies. The binding affinity was expressed as the binding score. The 2D and 3D structures were obtained from UCSF Chimera version 1.14 (Pettersen et al., 2004) and Accelrys Discovery Studio Visualizer (Dassault Systèmes BIOVIA 2017).
2.2 Molecular dynamic simulationThe shortlisted compound/ligand and the standard allantodapsone were studied further by molecular dynamics simulation within a Linux environment created by the Desmond modules of the Schrodinger (Schrödinger release maestro, 2021-4, 2021). The simple point charge water box solvent model in addition to the OPLS2005 force field was employed for the docking complexes made from protein-ligand interactions (Jorgensen and Tirado-Rives, 1988). To create a physiological environment, Na+ and Cl− ions were added for neutralization, which was followed by the addition of 0.15 M NaCl solution. The system was equilibrated by running NVT (number, volume, temperature) and NPT (number, pressure, temperature) ensembles for 100 ns and 12 ns, respectively (Martyna et al., 1992; Martyna et al., 1994). The simulation interaction diagram was generated by using the Desmond simulation interaction diagram tool of Maestro.
The root mean square deviation (RMSD), root mean square fluctuation (RMSF), and hydrogen bonds were estimated for the validation of the results. All the simulations were run in a set of three cycles for both the selected compound and the standard.
3 ResultThe basis of structure-based drug design is based on the identification of compounds by docking mechanisms and subsequent study of their molecular interactions via simulation.
3.1 Molecular docking to select compounds/ligands that bind to ClfA and ClfBThe docking results revealed a total of 5 compounds with the best binding energies for the ClfA and ClfB proteins. We also evaluated the binding energy of allantodapsone to compare it with that of the standard identified or shortlisted compounds. The binding energy of each shortlisted compound is given in Table 1 and Figure 1.
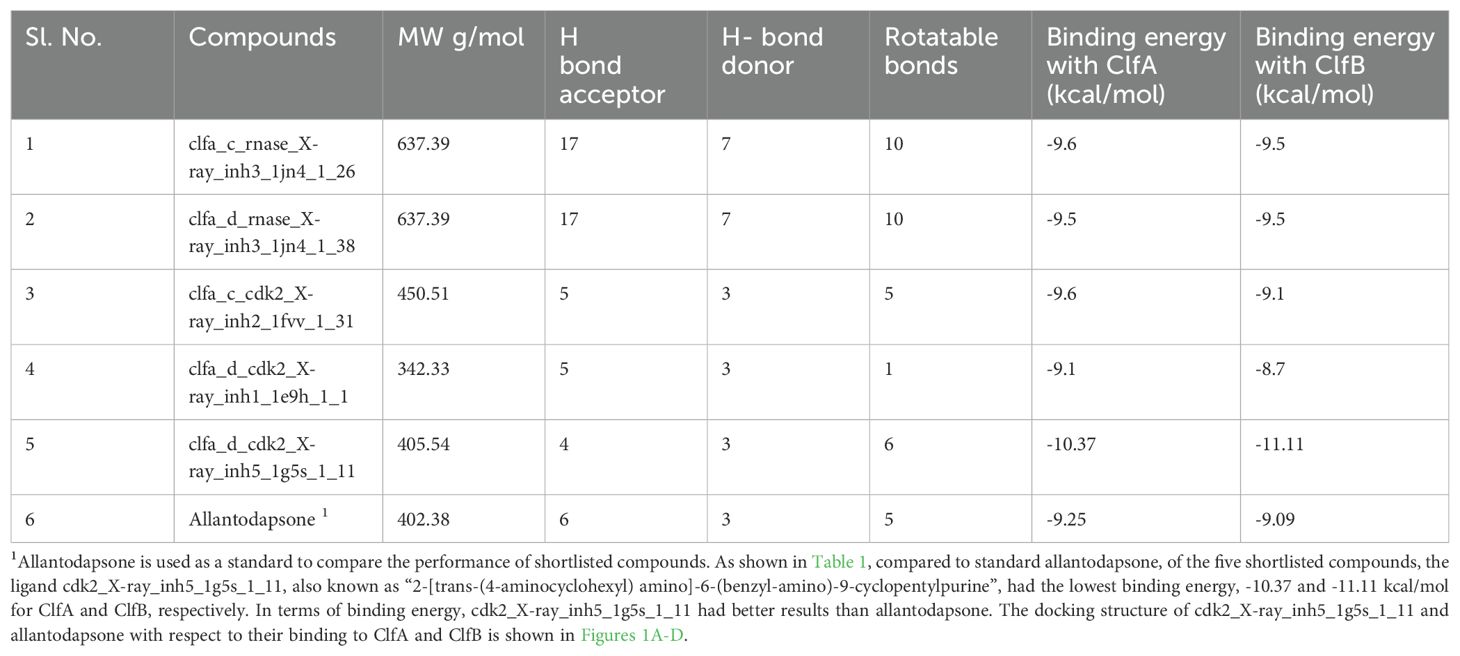
Table 1. Binding energy and other important properties of the shortlisted compounds for ClfA and ClfB.
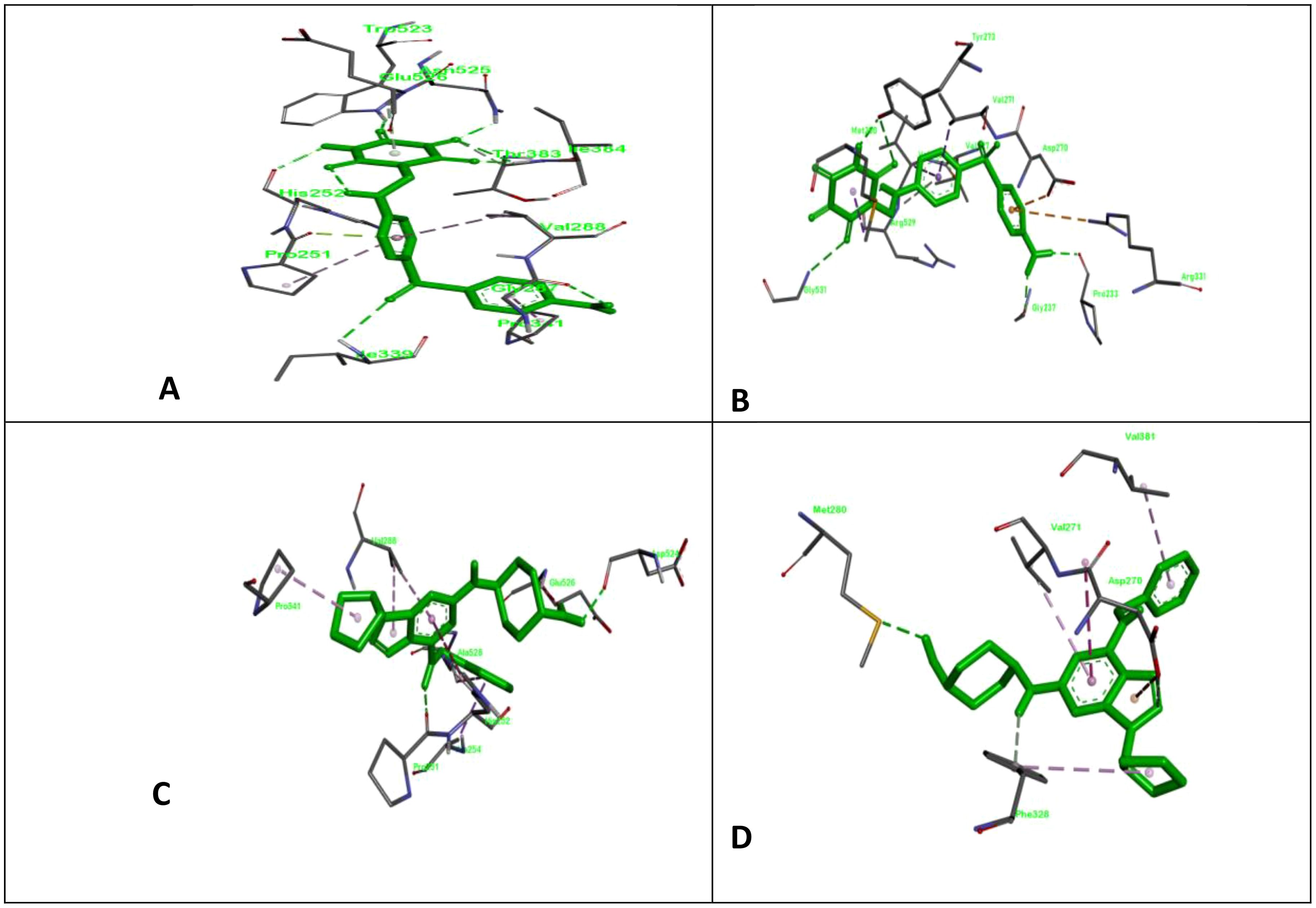
Figure 1. ClfA and ClfB are two components of the clumping factor adhesin protein that participate in ligand adhesin binding. Here, we show the docking complex of ClfA and B with standard and test compounds; (A) the docking complex of allantodapsone with ClfA; (B) the docking complex of allantodapsone with ClfB; (C) the docking complex of 2-[trans-(4-aminocyclohexyl) amino]-6-(benzyl-amino)-9-cyclopentylpurine with ClfA; and (D) the docking complex of 2-[trans-(4-aminocyclohexyl) amino]-6-(benzyl-amino)-9-cyclopentylpurine with ClfB.
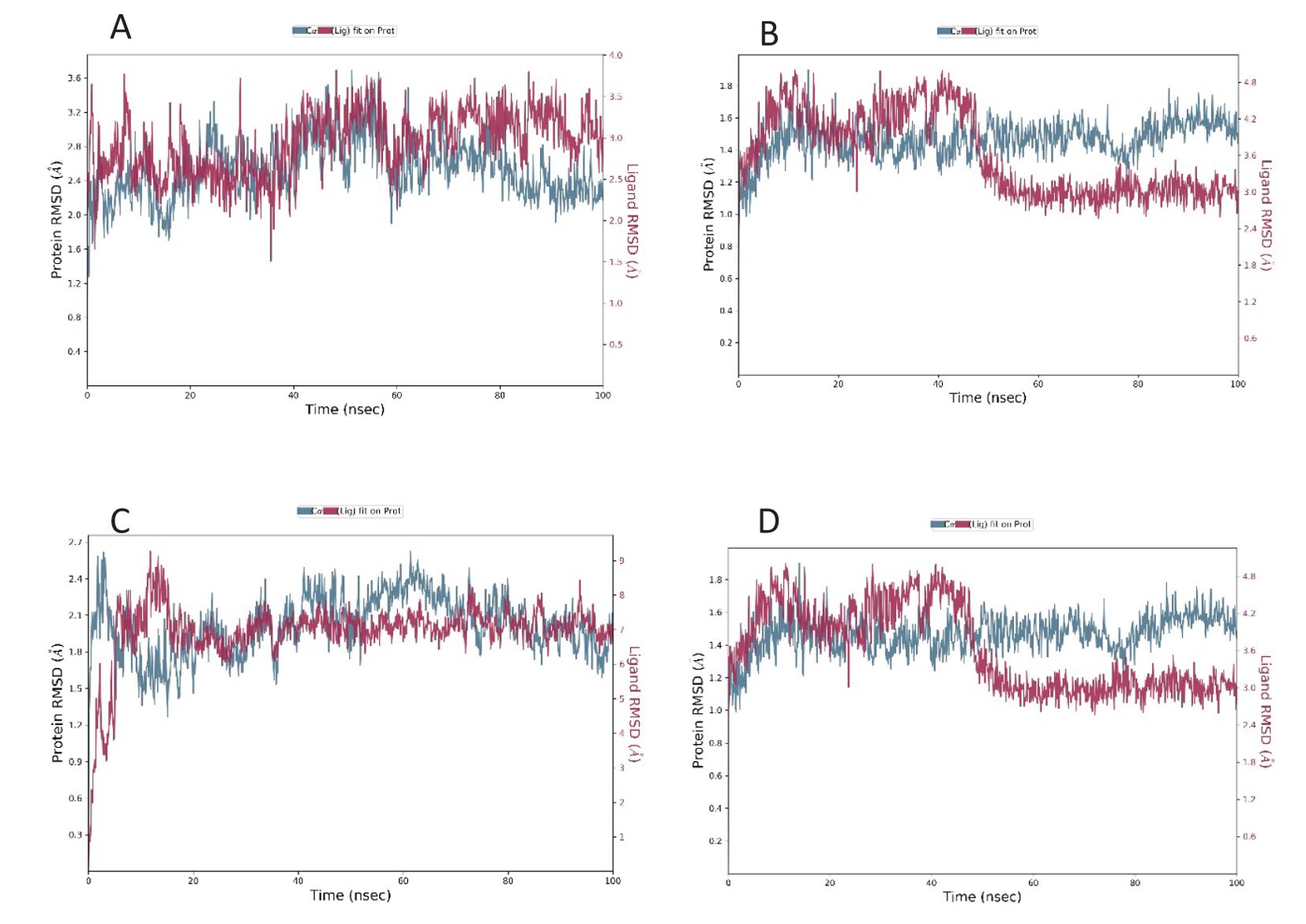
Figure 2. RMSD plot of the docked complex [Standard drug with ClfA (A) and ClfB (B)] [Candidate molecule with ClfA (C) and ClfB (D)].
3.2 Drug likeness and toxicity analysis of the selected compoundsThe drug likeness and toxicity of the selected compound 2-[trans-(4-aminocyclohexyl) amino]-6-(benzyl-amino)-9-cyclopentylpurine] were determined based on the outcome of various standard ADMET parameters. ADMET signifies the absorption of the drug, distribution of the drug, metabolism of the drug, excretion of the drug and toxicity of the drug. The important details of the ADMET properties of the test compounds are summarized in Tables 2, 3.
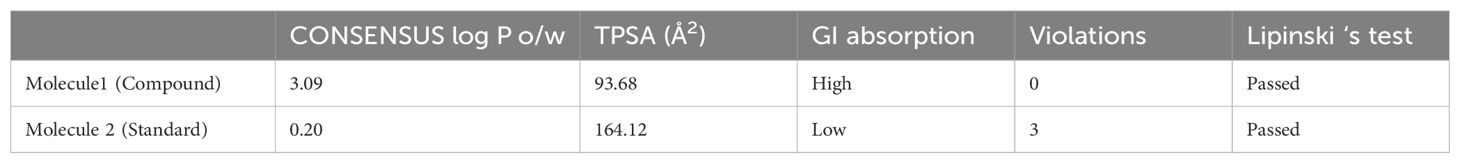
Table 2. Drug-likeness properties of the test compounds compared to those of standards.

Table 3. Drug toxicity status of the test compound.
3.3 Molecular simulation analysis of adhesin ligand interactionsThe interactions of the docking complex of the shortlisted compound and standard with ClfA and ClfB were further studied by molecular dynamics simulation.
The root mean square deviation was determined for the interaction of the compound as well as the standard with ClfA and ClfB. The average RMSD of the standard drug allantodapsone for binding ClfA was 1.76 Å, and that for binding to ClfB was 1.17 Å. The average RMSD of the candidate compound/ligand was 1.66 Å and 1.16 Å for the interaction of the standard drug allantodapsone with ClfA and ClfB, respectively (Figure 2). An RMSD less than 3 Å is considered a good interaction.
The root mean square fluctuation is a measure of residual fluctuation in a comparative study. The standard showed a peak at the 250th residue in the case of ClfA, while the candidate compound dissipated in areas closer to the 250th residue. In the case of ClfB, the standard as well as the candidate compound showed a peak closer to the 210th residue (Figure 3).
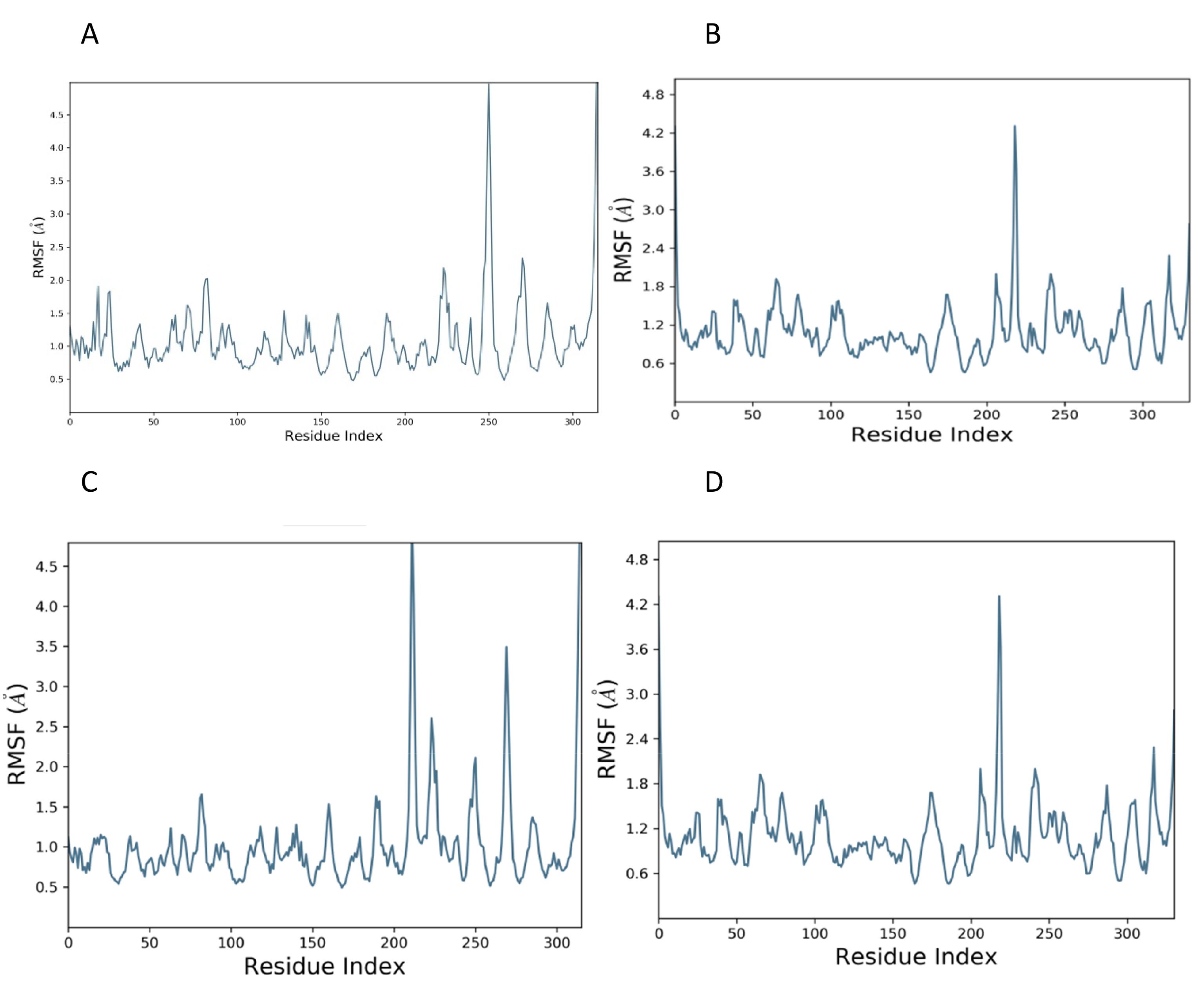
Figure 3. RMSF plot of docked complex [Standard drug with ClfA (A) and ClfB (B)]; [Candidate molecule with ClfA (C) and ClfB (D).
Protein–ligand interactions include four types of bonds: ionic, hydrophobic, waterbridge and hydrogen bonds. The majority of the interactions were waterbridge interactions. There were fewer ionic and hydrophobic interactions. Hydrogen bonds were detected between PRO, ASP and GLU in ClfA and between PRO, HIS, GLN, GLU, ASP and ASN in ClfB.
4 DiscussionAmong the traditional and rational methods of drug design, the latter is known to be more cost effective and efficient. It is also known to be based on the principle of reverse pharmacology, where an efficient protein target is identified and then molecules from a library are selected based on their interaction with the protein (Swinney and Anthony, 2011). In our study, we accomplished structure-based drug design by adopting molecular docking and molecular dynamics, which are the most common and reliable methods used. These methods help in understanding ligand–protein interactions, conformational changes, binding energy, etc (Kalyaanamoorthy and Chen, 2011). As a result of growing antibiotic resistance among Staphylococcus aureus due to overuse or misuse, there is an urgent need for the improvement of therapies for treating and controlling infection (Liu et al., 2020; Qureshi et al., 2023). Currently available antibacterial agents for MRSA include vancomycin, linezolid, clindamycin,daptomycin, dalbavancin, oritavancin, telavancin, etc; unfortunately, resistance to these drugs has already been reported worldwide. These drugs may no longer be reliable options for treating MRSA infections. Given these facts, the World Health Organization has included MRSA in the list of six high-priority pathogens that adversely affect public health (Willyard, 2017).
In recent years, many studies in which structure-based drug design has been attempted for the discovery of newer drugs to treat MRSA or staphylococcal infections have been reported. Ye et al., 2022, identified a compound that blocks the transcription of bacterial DNA. The compound worked by inhibiting transcription factors (Ye et al., 2022). Liu et al., 2020, targeted PVL and α-toxin with a compound named n-tetradecylphosphocholine (C14PC), which inhibits the binding of these virulence toxins to the cell through membrane phosphatidylcholine (Liu et al., 2020). Wang et al., 2018, reported a Sortase B (SrtB) inhibitor, Coptisine, a natural compound that does not exhibit antibacterial activity but can inhibit SrtB activity in vitro. In India, Morris et al., 2024, targeted amidase activity by designing the inhibitors SPECS-1 and SPECS-2, which reduced the biofilm-forming activity of bacteria (Morris et al., 2024). Zeenat et al., 2023, reported that the phytochemicals present in curcumin and eugenol are inhibitors of Eap (extracellular protein) homologs 1 and 2. It was tested and validated by molecular docking and dynamics (Zeenat et al., 2023). Rahman & Das (2021) concluded that surface adhesins are very promising as targets for designing vaccines. They identified 53 drug targets that are eligible for vaccine or drug development (Rahman and Das, 2021). Saha & Ghosh, 2023, targeted AgrA, a virulence factor regulator, to develop ligands that inhibit the binding of AgrA to its promoter site (Saha and Ghosh, 2023). Vijayakumar et al., 2022, tested the antibiofilm-forming ability of hesperidin, which is found in citrus fruits. They performed molecular docking to determine the interaction of hesperidin with the biofilm-forming SarA and CrtM proteins (Vijayakumar et al., 2022). Singh et al., 2022, targeted the protein and enzyme saDHFR, which is an important factor for the survival of bacteria (Singh et al., 2022). Sharma et al., 2024, identified inhibitors of crtM and sarA compounds from essential oils. crtM and sarA are proteins involved in biofilm formation (Sharma et al., 2024).
Riaz et al., 2023, in Pakistan, tested the binding affinity of three derivatives of quinolones, namely, sulfanilamide, 4-aminobenzoic acid, and sulfanilic acid, for five bacterial proteins. They found 2 out of 3 derivatives tested to be inhibitors of MRSA (Riaz et al., 2023). Prencipe et al., 2022, also developed the ClfA and B inhibitor allantodapsone, which has been found to have anti-colonizing properties in vitro (Prencipe et al., 2022). The present study also investigated the same adhesin proteins, ClfA and ClfB, to develop a ligand compound that interferes with the binding of ClfA and B to the host surface. We used allantodapsone as a standard in our study to validate and compare the findings. By comparison of various parameters, we found that the binding energies of the identified compounds with ClfA and ClfB (-10.37 and -11.11 kcal/mol, respectively) were greater than those of allantodapsone with ClfA and ClfB (-9.25 and -9.09 kcal/mol, respectively). The molecular dynamics results revealed that the interaction of the identified compound with ClfA and ClfB (1.66 Å and 1.16 Å, respectively) was greater than that of the control.
In silico identification of ligands and compounds that inhibit bacterial pathogenesis is an advantage for healthcare systems. Because this compound will target and avoid the colonization, an infection will not be established and development of resistance by the bacteria will not take place. Most of the studies that have been conducted on this topic have not been performed at any preclinical or clinical stage. The findings of in silico compatibility or interactions provide a foundation for in vitro and in vivo interactions with actual pathogens. The identified compound must show affinity for its target both in vitro and in vivo during the preclinical and clinical testing stages (Vijayalakshmi et al., 2014; Riaz et al., 2023). Prencipe et al. concluded that allantodapsone was capable of inhibiting the binding of bacteria to fibrinogen, loricrin and Ck10 components. Due to the shared ligand fibrinogen, allantodapsone was also able to block the interaction of FnBPA with fibrinogen. However, platelet aggregation and agglutination of S. aureus were not inhibited in the case of soluble fibrinogen (Prencipe et al., 2022). As mentioned earlier, our study also included ClfA and ClfB adhesins to identify the selected compound. Based on the findings of molecular docking and dynamics, the identified compound is an ideal inhibitor of Clf A and Clf B. Further in vitro and in vivo studies will confirm its role as a promising drug to prevent colonization and infections.
5 ConclusionStaphylococcus aureus is an important pathogen that is currently responsible for the high mortality rate in hospitals and in communities. Its pathogenicity is not only limited to humans but also widely observed in animals. MRSA is the most common resistant form of this organism. Due to increasing resistance to existing antibiotics, treatment has become a challenge, especially in patients with comorbidities or in the ICU. Designing a drug based on the target protein structure/active site and predetermined interaction and its dynamics can help in formulating a drug that is long lasting and promising.
Data availability statementThe original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributionsSS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing, Project administration, Resources, Software, Supervision, Validation. MB: Conceptualization, Investigation, Methodology, Resources, Software, Validation, Writing – original draft, Writing – review & editing. BU: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Writing – original draft, Writing – review & editing. SA: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Writing – original draft, Writing – review & editing. SF: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Writing – original draft, Writing – review & editing.
FundingThe authors declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Researchers Supporting project number (RSPD2024R966), King Saud University, Riyadh, Saudi Arabia, and Assam down town University, Guwahati, Assam, India.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References(2021). Schrödinger release maestro, 2021-4 (Schrödinger, LLC).
Banerjee, P., Eckert, A. O., Schrey, A. K., Preissner, R. (2018). ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263. doi: 10.1093/nar/gky318
PubMed Abstract | Crossref Full Text | Google Scholar
Berman, D. S., Eisner, W., Kreiswirth, B. (1993). Community-acquired methicillin-resistant staphylococcus aureus infection. New Engl. J. Med. 329, 1896–1896. doi: 10.1056/NEJM199312163292517
Crossref Full Text | Google Scholar
DeLeo, F. R., Otto, M., Kreiswirth, B. N., Chambers, H. F. (2010). Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375, 1557–1568. doi: 10.1016/S0140-6736(09)61999-1
PubMed Abstract | Crossref Full Text | Google Scholar
Hawkins, J., Kodali, S., Matsuka, Y. V., McNeil, L. K., Mininni, T., Scully, I. L., et al. (2012). A recombinant clumping factor a-containing vaccine induces functional antibodies to staphylococcus aureus that are not observed after natural exposure. Clin. Vaccine Immunol. 19, 1641–1650. doi: 10.1128/CVI.00354-12
PubMed Abstract | Crossref Full Text | Google Scholar
Jarvis, W. R., Schlosser, J., Chinn, R. Y., Tweeten, S., Jackson, M. (2007). National prevalence of methicillin-resistant Staphylococcus aureus in inpatients at US health care facilities, 2006. Am. J. infection control 35, 631–637. doi: 10.1016/j.ajic.2007.10.009
Crossref Full Text | Google Scholar
Jorgensen, W. L., Tirado-Rives, J. (1988). The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666. doi: 10.1021/ja00214a001
PubMed Abstract | Crossref Full Text | Google Scholar
Josse, J., Laurent, F., Diot, A. (2017). Staphylococcal adhesion and host cell invasion: Fibronectin-binding and other mechanisms. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02433
PubMed Abstract | Crossref Full Text | Google Scholar
Klein, E., Smith, D. L., Laxminarayan, R. (2007). Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999-2005. Emerging Infect. Dis. 13, 1840–1846. doi: 10.3201/eid1312.070629
Crossref Full Text | Google Scholar
Klevens, R. M., Edwards, J. R., Gaynes, R. P., National Nosocomial Infections Surveillance System (2008). The impact of antimicrobial-resistant, health care-associated infections on mortality in the United States. Clin. Infect. diseases: an Off. Publ. Infect. Dis. Soc. America 47, 927–930. doi: 10.1086/591698
Crossref Full Text | Google Scholar
Liu, J., Kozhaya, L., Torres, V. J., Unutmaz, D., Lu, M. (2020). Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence. J. Biol. Chem. 295, 5944–5959. doi: 10.1074/jbc.RA120.012697
PubMed Abstract | Crossref Full Text | Google Scholar
Martyna, G. J., Klein, M. L., Tuckerman, M. (1992). Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643. doi: 10.1063/1.463940
Crossref Full Text | Google Scholar
Martyna, G. J., Tobias, D. J., Klein, M. L. (1994). Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189. doi: 10.1063/1.467468
Crossref Full Text | Google Scholar
Moore, T. J., Zhang, H., Anderson, G., Alexander, G. C. (2018). Estimated costs of pivotal trials for novel therapeutic agents approved by the US food and drug administration, 2015-2016. JAMA Internal Med. 178, 1451–1457. doi: 10.1001/jamainternmed.2018.3931
Crossref Full Text | Google Scholar
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi: 10.1002/jcc.21256
PubMed Abstract | Crossref Full Text | Google Scholar
Morris, S. D., Kumar, V. A., Biswas, R., Mohan, C. G. (2024). Identification of a Staphylococcus aureus amidase catalytic domain inhibitor to prevent biofilm formation by sequential virtual screening, molecular dynamics simulation and biological evaluation. Int. J. Biol. Macromolecules 254, 127842. doi: 10.1016/j.ijbiomac.2023.127842
Crossref Full Text | Google Scholar
Mulcahy, M. E., Geoghegan, J. A., Monk, I. R., O’Keeffe, K. M., Walsh, E. J., Foster, T. J., et al. (2012). Nasal colonization by Staphylococcus aureus depends upon clumping factor B binding to the squamous epithelial cell envelope protein loricrin. PloS Pathog. 8, e1003092. doi: 10.1371/journal.ppat.1003092
PubMed Abstract | Crossref Full Text | Google Scholar
Otto, M. (2010). Basis of virulence in community-associated methicillin-resistant staphylococcus aureus. Annu. Rev. Microbiol. 64, 143–162. doi: 10.1146/annurev.micro.112408.134309
PubMed Abstract | Crossref Full Text | Google Scholar
Paharik, A. E., Horswill, A. R. (2016). The staphylococcal biofilm: Adhesins, regulation, and host response. Microbiol. Spectr. 4, 4.2.06. doi: 10.1128/microbiolspec.VMBF-0022-2015
Crossref Full Text | Google Scholar
Patti, J. M., Allen, B. L., McGavin, M. J., Höök, M. (1994). MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48, 585–617. doi: 10.1146/annurev.mi.48.100194.003101
PubMed Abstract | Crossref Full Text | Google Scholar
Peacock, S. J., Moore, C. E., Justice, A., Kantzanou, M., Story, L., Mackie, K., et al. (2002). Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infection Immun. 70, 4987–4996. doi: 10.1128/IAI.70.9.4987-4996.2002
Crossref Full Text | Google Scholar
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
PubMed Abstract | Crossref Full Text | Google Scholar
Prencipe, F., Alsibaee, A., Khaddem, Z., Norton, P., Towell, A. M., Ali, A. F. M., et al. (2022). Allantodapsone is a pan-inhibitor of staphylococcus aureus adhesion to fibrinogen, loricrin, and cytokeratin 10. Microbiol. Spectr. 10, e0117521. doi: 10.1128/spectrum.01175-21
PubMed Abstract | Crossref Full Text | Google Scholar
Qureshi, B., Khalil, R., Saeed, M., Nur-e-Alam, M., Ahmed, S., Ul-Haq, Z. (2023). Structure-based discovery of potent staphylococcus aureus thymidylate kinase inhibitors by virtual screening. Medicinal Chem. 19, 75–90. doi: 10.2174/1573406418666220407092638
Crossref Full Text | Google Scholar
Rahman, S., Das, A. K. (2021). Integrated multiomics, virtual screening and molecular docking analysis of methicillin-resistant staphylococcus aureus usa300 for the identification of potential therapeutic targets: An in-silico approach. Int. J. Pept. Res. Ther. 27, 2735–2755. doi: 10.1007/s10989-021-10287-9
PubMed Abstract | Crossref Full Text | Google Scholar
Riaz, F., Hossain, M., Roney, M., Ali, Y., Qureshi, S., Muhammad, R., et al. (2023). Evaluation of potential bacterial protease inhibitor properties of selected hydroxyquinoline derivatives: An in silico docking and molecular dynamics simulation approach. J. Biomolecular Structure Dynamics 41, 9756–9769. doi: 10.1080/07391102.2022.2146200
Crossref Full Text | Google Scholar
Saha, S., Ghosh, M. (2023). Computational exploration of natural compounds targeting Staphylococcus aureus: Inhibiting AgrA promoter binding for antimicrobial intervention. J. Biomolecular Structure Dynamics, 1–12. doi: 10.1080/07391102.2023.2246566
Crossref Full Text | Google Scholar
Sampedro, G. R., Bubeck Wardenburg, J. (2017). Staphylococcus aureus in the intensive care unit: are these golden grapes ripe for a new approach? J. Infect. Dis. 215, S64–S70. doi: 10.1093/infdis/jiw581
PubMed Abstract | Crossref Full Text | Google Scholar
Sharma, V., Gogoi, B., Borah, S. N., Ghosh, A., Mazumdar, A., Kalita, R. D. (2024). in-silico molecular docking and molecular dynamic simulation of γ-elemene and caryophyllene identified from the essential oil of kaempferia galanga l. Against biofilm forming proteins, crtm and sara of staphylococcus aureus. J. Biomolecular Structure Dynamics, 1–13. doi: 10.1080/07391102.2024.2310773
Crossref Full Text | Google Scholar
Singh, G., Soni, H., Tandon, S., Kumar, V., Babu, G., Gupta, V., et al. (2022). Identification of natural DHFR inhibitors in MRSA strains: Structure-based drug design study. The results. Chem. 4, 100292. doi: 10.1016/j.rechem.2022.100292
Crossref Full Text | Google Scholar
Soltani, E., Farrokhi, E., Zamanzad, B., Shahini Shams Abadi, M., Deris, F., Soltani, A., et al. (2019). Prevalence and distribution of adhesins and the expression of fibronectin-binding protein (FnbA and FnbB) among Staphylococcus aureus isolates from Shahrekord Hospitals. BMC Res. Notes 12, 49. doi: 10.1186/s13104-019-4055-0
PubMed Abstract | Crossref Full Text | Google Scholar
Swinney, D. C., Anthony, J. (2011). How were new medicines discovered? Nat. Rev. Drug Discovery 10, 507–519. doi: 10.1038/nrd3480
Crossref Full Text | Google Scholar
Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L., Fowler, V. G. J. R. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661. doi: 10.1128/CMR.00134-14
PubMed Abstract | Crossref Full Text | Google Scholar
Troeman, D. P. R., Hazard, D., Timbermont, L., Malhotra-Kumar, S., Van Werkhoven, C. H., Wolkewitz, M., et al. (2023). Postoperative staphylococcus aureus infections in patients with and without preoperative colonization. JAMA Network Open 6, e2339793. doi: 10.1001/jamanetworkopen.2023.39793
PubMed Abstract | Crossref Full Text | Google Scholar
Vijayakumar, K., Muhilvannan, S., Arun Vignesh, M. (2022). Hesperidin inhibits biofilm formation, virulence and staphyloxanthin synthesis in methicillin resistant Staphylococcus aureus by targeting SarA and CrtM: An in vitro and in silico approach. World J. Microbiol. Biotechnol. 38, 44. doi: 10.1007/s11274-022-03232-5
留言 (0)