Patient tissue specimens and cell lines
Tissue from CRC patients were obtained from the First Affiliated Hospital of Zhengzhou University. A CRC tissue cDNA chip was obtained from Shanghai Outdo Biotech (Shanghai, China). All patients signed informed consent forms, and the protocols were approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University (2019-KY-423) and Shanghai Outdo Biotech (YB M-05-02). HCT116 cells were obtained from the Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China). SW480 cells and Human embryonic kidney 293T cells (293T cells) were generous gifts from the Biotherapy Center of the First Affiliated Hospital of Zhengzhou University. HCT116 cells and SW480 cells were cultured in high-glucose DMEM (Gibco, Carlsbad, CA, USA), 293T cells were cultured in RPMI 1640 (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Clark Bioscience, Richmond, VA, USA) at 37 ℃ and 5% CO2.
M7G-RIP sequencing
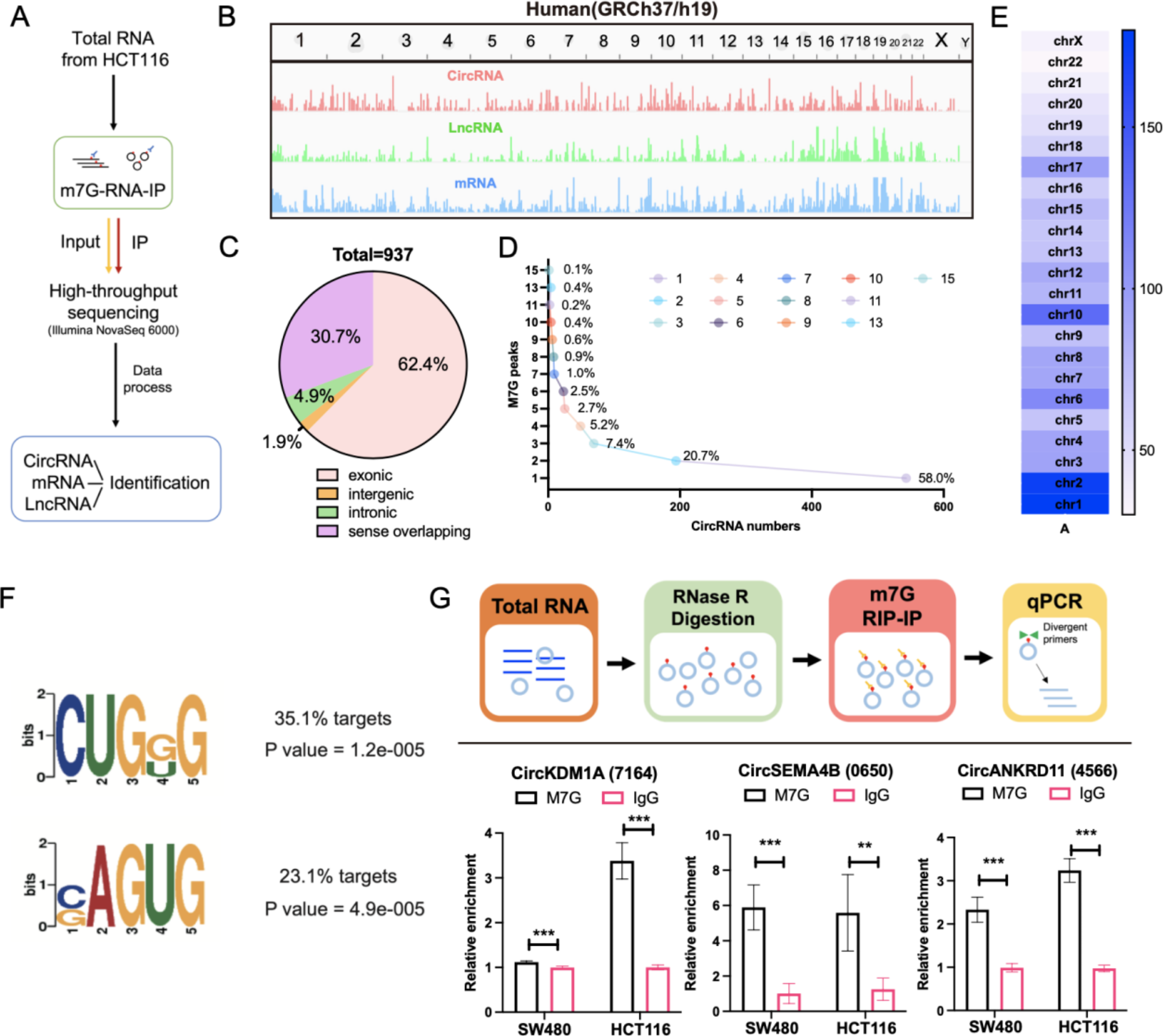
The m7G-IP-Seq service was provided by CloudSeq Inc. (Shanghai, China). Total RNA from HCT116 cells was collected and processed using RNase R digestion. RNase R, an exonuclease, recognizes and binds to the 3’ end of RNA molecules to degrade linear RNA and enrich circRNA, which lacks free 3’ or 5’ ends due to its closed loop structure. mRNA was then further isolated using oligo(dT) magnetic beads (ThermoFisher). Immunoprecipitation was performed according to the instructions of GenSeqTM m7G-IP kit (GenSeq Inc., China). RNA libraries for IP and input samples were then constructed with NEBNext® Ultra II Directional RNA Library Prep Kit (New England Biolabs, Inc., USA) by following the manufacturer’s instructions. Libraries were qualified using Agilent 2100 bioanalyzer and then sequenced in a NovaSeq platform (Illumina). Briefly, Paired-end reads were harvested from Illumina NovaSeq 6000 sequencer, and were quality controlled by Q30. After 3’adaptor-trimming and low-quality reads removing by cutadapt software (v1.9.3). First, clean reads of input libraries were aligned to reference genome (UCSC HG38) by STAR software. Then circRNAs were identified by DCC software using the STAR alignment results. After that, clean reads of all libraries were aligned to the reference genome by Hisat2 software (v2.0.4). Methylated sites on RNAs (peaks) were identified by MACS software. Differentially methylated sites were identified by diffReps. These peaks identified by both softwares overlapping with exons of mRNA, LncRNA and circRNA were figured out and chosen by home-made scripts. GO and Pathway enrichment analysis were performed by the differentially methylated protein coding genes, the associated genes of differentially methylated LncRNAs and the source genes of differentially methylated circRNAs separately.
SiMETTL1 sequencing
The SiMETTL1-seq service was provided by CloudSeq (Shanghai, China). SiRNAs targeting the backsplice junction site of METTL1 were synthesized by RiboBio (RiboBio, Guangzhou, China). Total RNA from HCT116 transfected si-METTL1 was purified using QIAGEN RNeasy Kit. Amplification was performed using the AffinityScript-RT kit using Cy3 labeling. After elution, raw images were obtained by scanning with an Agilent Scanner G5761A (Agilent Technologies). Feature Extraction software (version12.0.3.1, Agilent Technologies) was used to process the original image to extract the original data. Quantile normalization and subsequent processing were performed with the use of Genespring software (version14.8, Agilent Technologies). The normalized data were filtered so that at least one set of probes 100% labeled as detected from each set of samples used for comparison was left for subsequent analysis. The p value of T test was used to screen differentially expressed genes. Then, GO and KEGG enrichment analysis were performed to determine the main biological functions or pathways affected by the differential genes. Finally, unsupervised hierarchical clustering was performed on the differentially expressed genes, and the expression patterns of differentially expressed genes among different samples were displayed in the form of heat maps.
SiRNA and plasmid construction
The full-length of circKDM1A was cloned into over expression vector pcDNA3.1 (Hanbio Biotechnology, Wuhan, China), while the mock vector with no circKDM1A sequence served as a control. SiRNAs targeting the backsplice junction site of circKDM1A and METTL1 were synthesized by RiboBio (RiboBio, Guangzhou, China). Moreover, the shMETTL1 were synthesized by GenePharma (GenePharma, Shanghai, China), efficiency detected by qRT-PCR. miR-147b-3p mimics were purchased from RiboBio (RiboBio, Guangzhou, China). The wild-type and mutant circKDM1A plasmids were synthesized by Genechem (Genechem, Shanghai, China). The wild-type and mutant METTL1 were synthesized by Hanbio (Hanbio, Shanghai, China) Lipofectamine 3000 (Invitrogen, Carlsbad, USA) were used to cell transfections. The sequences of siRNAs were listed in Table S1.
RNA isolation, reverse transcription, and qRT-PCR
Total RNA was isolated from cells and tissues using RNAiso Plus (Takara, Dalian, China) following the manufacturer’s instructions. The integrity and purity of the extracted total RNA were determined using NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham, USA). RNA was immediately stored at -80 °C until use. Reverse transcription was carried out using the All-in-One™ First-Strand cDNA Synthesis Kit (Uelandy, Suzhou, China). Genomic DNA was removed at 42 °C for 2 min. Subsequently, RNA from cells and tissues was reverse-transcribed into cDNA at 37 °C for 15 min, followed by an inactivation step at 85 °C for 5 s. qRT-PCR was performed using the QuantStudio 5 Real-Time PCR System (Applied Biosystems, Foster City, USA) and the Hieff qPCR SYBR Green Master Mix kit (Yeasen, Shanghai, China). The qRT-PCR reactions were conducted with an initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and annealing at 60 °C for 30 s. Primer sequences used for qRT-PCR are listed in Table S2. The relative quantification of RNA was calculated using the 2^−ΔΔCt method, with GAPDH as the internal reference.
Western blot analysis
Total protein was obtained by lysis of cells and centrifugation of tissue samples using RIPA buffer containing protease and phosphatase inhibitors on ice and homogenized with a BCA Kit (Beyotime, China). Denaturation by heat. SDS-PAGE gels were assembled in appropriate percentages based on the expected protein size range. The gel was run at a constant voltage of 100 V until the protein was sufficiently separated. The separated proteins were transferred from the gel to the PVDF membrane (Millipore, Massachusetts, USA) using a wet transfer system. 5% skim milk powder in 1xTBST was blocked at room temperature. The membranes were incubated overnight at 4℃ with the following primary antibodies: AKT (ab179463) from Abcam (MA, USA), anti-phospho-AKT (Ser473) (4060), anti-PDK1 (3062) and anti-E-cadherin (3195) from Cell Signaling Technology (MA, USA), anti-Palladin (No. 10853-1-AP), and anti-GAPDH (No. 10494-1-AP) from Proteintech (Wuhan, China). Membranes were incubated for 1 h in blocking buffer with a secondary antibody attached to HRP. Enhanced chemiluminescence reagents (Millipore, MA, USA) were used to visualize the protein bands. Protein band signals were captured using a chemiluminescence imaging system.
CircRNA identification
Total RNA and gDNA was extracted from CRC cells. Forward and reverse primers based on the specific junction sequence of the circular RNA for circKDM1A are designed to amplify the sequences in both cDNA and gDNA. The presence and specificity of the amplification products are validated using qRT-PCR and nucleic acid electrophoresis.
RNA Stability Assay: Cells are treated with 100 ng/mL actinomycin D (Merck, Darmstadt, Germany), and total RNA is harvested and extracted at 0 h, 8 h, 16 h, and 24 h post-treatment. The changes in circRNA expression levels are quantified using qRT-PCR.
Fluorescence in situ hybridization (FISH)
12 mm diameter coverslips are placed at the bottom of a 24-well plate, and an appropriate number of cells (6 × 10^4 cells/well) are cultured overnight. The cells are allowed to reach 60–70% confluency before the experiment. At room temperature, the cells are fixed with 4% paraformaldehyde in PBS for 20 min. The specific Cy3-labeled circKDM1A probe (Table S1) (GenePharma, Shanghai, China) is hybridized using a FISH kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions. Briefly, pre-hybridization is performed at 37 °C for 30 min, followed by hybridization with 2.5 μL of 20 μM circKDM1A probe at 37 °C overnight. The cells are then stained with DAPI. Image acquisition is carried out using a laser scanning confocal microscope (Zeiss, Jena, Germany).
Immunohistochemistry (IHC) and paraffin –SweAMI -FISH + double IF
Paraffin sections were baked for 2 h, after deparaffinization, microwave repair cooling, permeabilization, and sealing, METTL1 primary antibody (No. 14994-1-AP, proteintech) or phospho-AKT primary antibody (GB150002-100, servicebio) was drip-added at low temperature overnight, and the signal was amplified by a DAB system (PV-6000D, Zsgb-bio, China). Imaged under a microscope (Olympus, Tokyo, Japan). A semi-quantitative ISH scoring criterion was used to assess METTL1 expression. METTL1 and phospho-AKTwere scored using Quant Center (3DHISTECH, Budapest, Hungary).
For Paraffin-SWEAMI-FISH + double IF, Paraffin sections were processed as above until antigen retrieval. The above FISH experimental procedures were completed. The cells were blocked by dropping BSA for 30 min. Primary antibody against METTL1 was added and incubated overnight at 4℃. Cy5-labeled secondary antibodies were incubated for 50 min at room temperature. The sections were blocked again and the PDK1 primary antibody was added dropfold at 4℃ overnight. Tsa-488-labeled secondary antibodies were incubated for 50 min at room temperature. DAPI was used to stain nuclei, and anti-fluorescence quenching sealing agent was added dropped-on to seal tablets. The sections were observed, and images were collected under a Nikon orthostatic fluorescence microscope.
RNA pull-down assay
The BersinBio™ RNA pulldown Kit (Bes5102) was used for the experiments. Biotinylated circKDM1A probe, biotinylated miR147b-3p probe, and their corresponding control probes were designed and synthesized by Genepharm (Shanghai, China) (Table S1). The RNA probes were heated to 95 °C for 2 min and then immediately placed on ice for 2 min to prepare RNA secondary structures. 40 μL of streptavidin magnetic beads were washed with NT2 buffer and then incubated with the RNA probes at 25 °C for 30 min. Cells were lysed in NT2 buffer containing protease inhibitors and RNase inhibitors, incubated on ice for 10 min, and then centrifuged. The supernatant was collected and incubated with the probe-bead complexes at 25 °C with gentle rotation for 2 h. The magnetic beads were collected and washed four times by adding 1 mL of NT2 buffer each time. Proteins were eluted by adding proteinase K and incubating at 55 °C for 30 min with intermittent mixing. RNA was extracted using the RNA Clean & Concentrator™-25 kit (R1017, ZYMO Research, USA) for subsequent qRT-PCR analysis.
RNA immunoprecipitation (RIP) assay
A total of 50 μl protein A/G beads were incubated with 5 μg (METTL1, AGO2) of the experimental antibody, or an equal amount of IgG antibody (Millipore, MA, USA) mixed at room temperature for 30 min. Cells were fully lysed using 1X RIP lysis buffer, and total cell lysates were mixed with the complexes described above and rotated overnight at 4℃. The beads were collected, and the supernatant was removed to add 1 ml 1x RIP wash buffer, and the beads were washed 6 times in total. Digestion was performed using proteinase k for 30 min. After digestion, RNA was purified using the RNA Clean & Concentrator™ kit (ZYMO Research, USA) to remove the enzyme and buffer components. The RNA was reverse transcribed using Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Interactions between METTL1, AGO2, and circKDM1A transcripts were assessed by qPCR and normalized to the input.
m7G-circRNA meRIP
Total RNA was extracted from 2 × 10^7 cells. The RNA (2 μg) was treated with RNase R (4 U) (Epicentre Biotechnologies, Madison, WI, USA) at 37℃ for 15 min to digest linear RNA. Protein A/G magnetic beads were blocked with PBS containing 5% BSA to prevent nonspecific binding. The beads were then coupled with the m7G antibody (RN017M, MBL) by rotating at room temperature for 1 h. RNA fragments from the RNase R digestion were incubated with the bead-antibody complexes at 4℃ for 4 h with gentle rotation. The beads were washed six times with 1x RIP wash buffer to remove nonspecifically bound RNA. The RNA/antibody complexes were digested with Proteinase K at 55℃for 30 min to remove proteins and release the RNA. After digestion, RNA was purified using the RNA Clean & Concentrator™ kit (ZYMO Research, USA) to remove the enzyme and buffer components. The RNA was reverse transcribed using Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). The enrichment of m7G-modified RNA was assessed by qRT-PCR and normalized to the input RNA.
Transwell assay
For cell migration assays, 5 × 105 modified HCT116 and SW480 cells were seeded in transwell chambers with an 8 μm well membrane (Corning, NY, USA). After 36–72 h of culture, the lower layer cells were fixed, and the upper layer cells were removed by wiping. Migrating cells were stained with Giemsa (Solarbio, Beijing, China). Images of the cells were obtained using light microscopy in four fields. Similarly, for invasion assays, 5 × 105 modified HCT116 and SW480 cells were seeded in transwell chambers with an 8 μm pore membrane (Corning, NY, USA) coated with Matrigel (Corning, NY, USA). The invasive cells were stained and imaged. The number of migrating cells and invasive cells were counted.
Edu and CCK8 assay
Edu experiments were performed with the support of Cell-Light TM Edu Apollo In Vitro Kit (C10310-1) (RiboBio, Guangzhou, China). According to the instructions, 2 × 103 modified HCT116 or SW480 cells were seeded in 96-well plates, the cells were cultured for 48 h, Edu labeling was added. Then, apollo staining and DNA staining was performed, and images of different channels were collected by fluorescence microscope (Olympus Corporation, Tokyo, Japan) and synthesized. For the CCK8 assay, 2000 modified HCT116 or SW480 cells were seeded in 96-well plates. After the cells were cultured for 0, 12, 24, 48, 72 and 96 h, the cells were added to the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) and incubated at 37 °C for 2 h. The OD450 value was measured using a microplate detector.
Colony formation and cell cycle and apoptosis
For the clone formation assay, 2 × 103 modified CRC cells were seeded in 12-well plates. After 4 weeks, the cells had grown in groups, they were fixed, Giemsa stained, and photographs were taken. The number of communities was counted using Image J (National Institutes of Health, Bethesda, MD, USA). The apoptosis of modified HCT116 or SW480 cells was detected by Annexin V/propidium iodide (PI) apoptosis detection kit (Beyotime, Shanghai, China). After the cells were collected, FITC/mCherry-Annexin V and PI double staining were added and detected by flow cytometry (ACEA NoVoCyte, USA). The cell cycle experiment was conducted using the Cell Cycle Detection Kit (C6031, Uelandy) as per the manufacturer’s instructions. Collected cells were fixed overnight in pre-cooled 70% ethanol. Following fixation, the cells were washed with PBS and stained with RNaseA and PI at room temperature for 30 min. Flow cytometry analysis was performed using the ACEA NovoCyte system (USA). The acquired data were analyzed and fitted using FlowJo™ software.
Animal models
EGFP-sh-METTL1, luci-lv-METTL1, mcherry-lv-circKDM1A, and mcherry-mut-circKDM1A plasmids, along with their respective control plasmids (Genepharm, Shanghai, China), were co-transfected with pSPAX2 and pMD2G plasmids into 293T cells for virus packaging. The concentrated viruses were then used to transduce HCT116 cells using LipoFilter Reagent (Hanbio Biotechnology, Wuhan, China). Stable cell lines were established by selecting with puromycin (2 μg/mL) or neomycin G418 (700 μg/mL). All mouse procedures were approved by the Institutional Animal Care and Use Committee of Zhengzhou University. BALB/c nude mice (4 weeks of age) were obtained from Vital River Laboratories. After a one-week acclimation period, the mice were injected via the tail vein with stable HCT116 cells (2 × 106 in 100 μL PBS) from each experimental group. The mice were regularly monitored for body weight and activity levels. The experiment was terminated if the mice showed significantly reduced activity, a body weight loss exceeding 20%, or severe cachexia. At the experimental endpoint, fluorescence was directly detected using the IVIS Illumina system (Caliper Life Sciences, USA), or bioluminescence was measured following intraperitoneal injection of D-luciferin (150 mg/kg, Yeasen, China). Mice from both the treatment and control groups were euthanized by overdose inhalation anesthesia. Lung metastases were harvested, fixed in paraffin, and sectioned for HE staining, immunohistochemistry, and immunofluorescence staining.
Statistical analysis
All data were analyzed using Prism 10.0 (GraphPad, San Diego, CA, USA) and expressed as mean ± SD. Chi-squared tests were performed using SPSS Statistics 21 (IBM, Chicago, IL, USA). Significant differences between two independent groups were evaluated by Student’s t-test. Correlations were analyzed using Pearson’s correlation coefficient(r) and two-tailed P-values. Survival curves were assessed by log-rank (Mantel-Cox) tests. p < 0.05 was considered significant. All experiments were performed at least three times.







留言 (0)